Download to read offline


















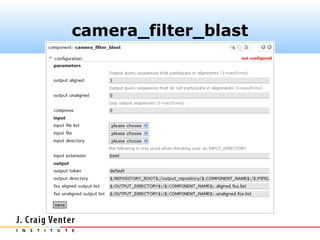
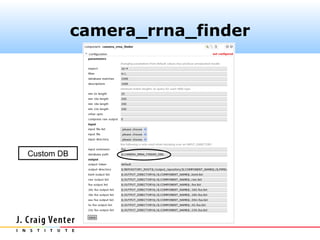

![rRNA Finder DB
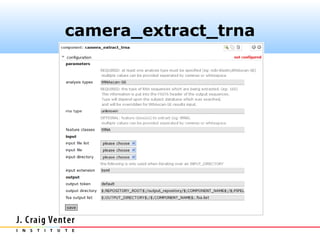
Fasta headers were coded as follows:
>#S [D] ...original.header...
where # is one of (5, 16, 18, 23) and D is one of
(A, B, E). The camera_rrna_finder
component expects this format.](https://image.slidesharecdn.com/cameraannotationpipeline-130410151041-phpapp02/85/CAMERA-metagenomic-annotation-pipeline-26-320.jpg)










![AnnotationData Objects
AnnotationData
AnnotationData::Polypeptide
Polypeptide
type:
[some string]
attributes: AnnotationData Object(s)
common_name
gene_symbol
EC
GO
TIGR_role
…](https://image.slidesharecdn.com/cameraannotationpipeline-130410151041-phpapp02/85/CAMERA-metagenomic-annotation-pipeline-45-320.jpg)








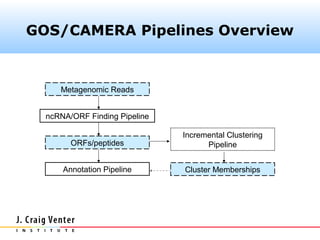
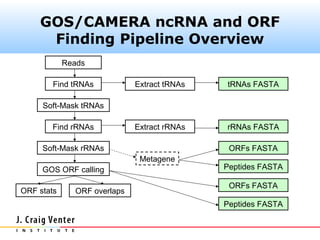
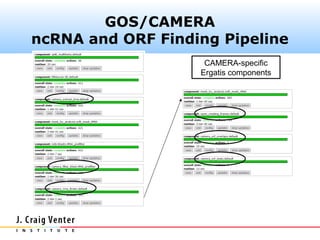
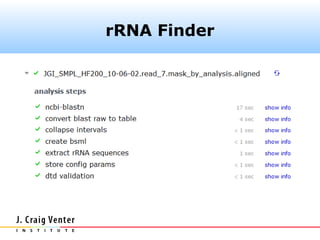
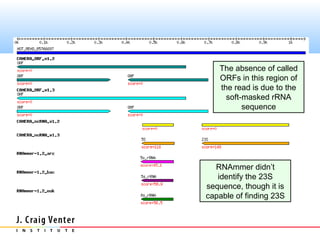
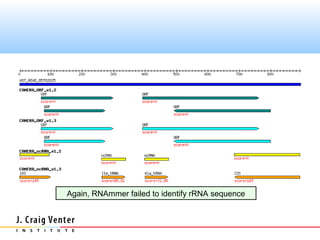
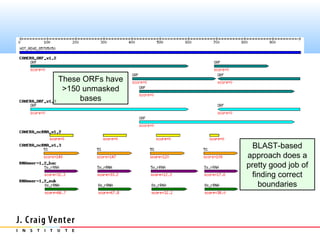
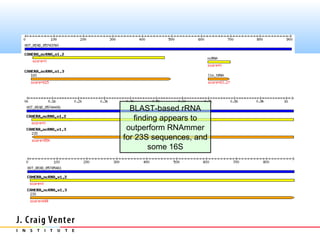
The document describes the CAMERA annotation pipelines and related infrastructure. It discusses the compute infrastructure including the CALIT2 compute grid and SOS cluster. It then describes the GOS/CAMERA ncRNA and ORF finding pipeline, including the rRNA finding pipeline, ORF calling, and tRNA extraction. It also discusses the GOS incremental protein clustering pipeline and the CAMERA annotation pipeline, including specifications and implementation. Finally, it provides thoughts on object-oriented design approaches for the annotation pipeline to support changing annotation rules and data sources over time.




![Writing an Ostinato Protocol Builder [FOSDEM 2021]](https://cdn.slidesharecdn.com/ss_thumbnails/fosdem-ostinato-210213055401-thumbnail.jpg?width=640&height=640&fit=bounds)











![[嵌入式系統] MCS-51 實驗 - 使用 IAR (2)](https://cdn.slidesharecdn.com/ss_thumbnails/mcs51iarpart2-150613071717-lva1-app6891-thumbnail.jpg?width=640&height=640&fit=bounds)


















































