Download to read offline




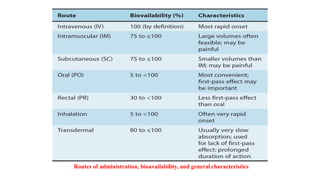
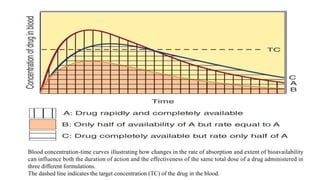

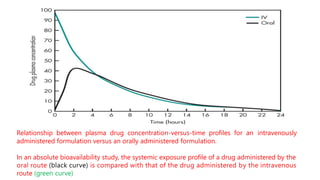
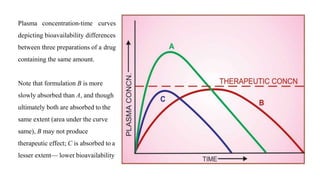











The lecture by Dr. Awais Irshad covers key pharmacology concepts such as bioavailability, bioequivalence, half-life, and drug dosing strategies. It details the factors affecting drug absorption and metabolism, including routes of administration, first-pass elimination, and physicochemical properties. Additionally, it discusses the significance of loading and maintenance doses in achieving effective drug concentrations in the plasma.





![Nonlinear pharmacokinetics.pptx1[1]](https://cdn.slidesharecdn.com/ss_thumbnails/nonlinearpharmacokinetics-201215071246-thumbnail.jpg?width=640&height=640&fit=bounds)















































![ABDOMINAL EXAMINATION Presentation[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/abdominalexaminationpresentation1-240105120242-b6318479-thumbnail.jpg?width=640&height=640&fit=bounds)




![Down syndrome (2)[1].pptx pediatric lecture](https://cdn.slidesharecdn.com/ss_thumbnails/downsyndrome21-240709094926-fcdd02d9-thumbnail.jpg?width=640&height=640&fit=bounds)




























