Download to read offline













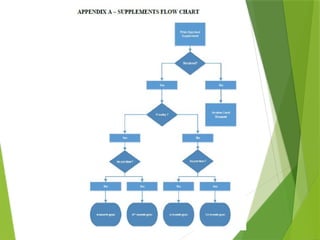


This document outlines the performance goals and requirements for submitting Prior Approval Supplements (PAS) under the GDUFA III agreement between the generic drug industry and the FDA. It categorizes changes to approved applications as major, moderate, or minor and specifies varying submission timelines and inspections based on these classifications. Additionally, it addresses grouped supplements, incorrect reporting categories, and the appeal process if an applicant disagrees with FDA's determinations.













































































