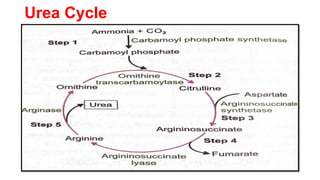
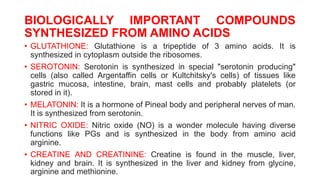
The document outlines the digestion, absorption, metabolism, and disorders of amino acids and proteins, detailing the biochemical processes involved in breaking down dietary proteins into amino acids. It also discusses various metabolic disorders resulting from enzyme deficiencies, such as phenylketonuria and alkaptonuria, and explains the causes of proteinuria and hypoproteinemia. Additionally, the document describes the synthesis of biologically important compounds from amino acids and the transport of ammonia resulting from metabolic processes.