Downloaded 51 times
![Advancing the Metagenomics Revolution Invited Talk Symposium #1816, Managing the Exaflood: Enhancing the Value of Networked Data for Science and Society San Diego, CA February 2010 Dr. Larry Smarr Director, California Institute for Telecommunications and Information Technology Harry E. Gruber Professor, Dept. of Computer Science and Engineering Jacobs School of Engineering, UCSD [email_address]](https://image.slidesharecdn.com/tcalit2widewebextranethtmlnewsroompresentationslsmarr2009pptaaasmetagenomics021910final-100219162443-phpapp02/85/Advancing-the-Metagenomics-Revolution-1-320.jpg)
![Advancing the Metagenomics Revolution Invited Talk Symposium #1816, Managing the Exaflood: Enhancing the Value of Networked Data for Science and Society San Diego, CA February 2010 Dr. Larry Smarr Director, California Institute for Telecommunications and Information Technology Harry E. Gruber Professor, Dept. of Computer Science and Engineering Jacobs School of Engineering, UCSD [email_address]](https://image.slidesharecdn.com/tcalit2widewebextranethtmlnewsroompresentationslsmarr2009pptaaasmetagenomics021910final-100219162443-phpapp02/75/Advancing-the-Metagenomics-Revolution-1-2048.jpg)

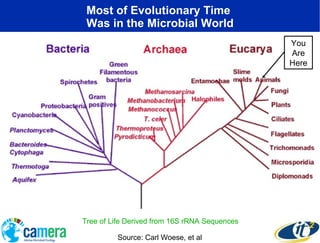





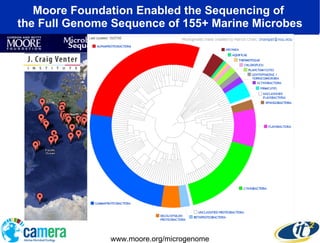


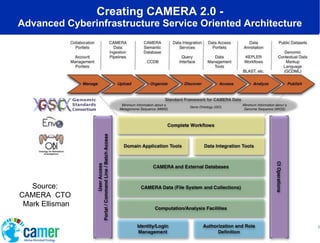
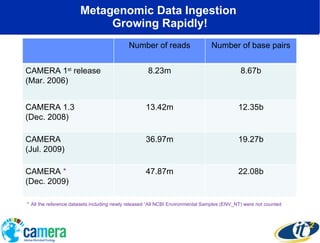
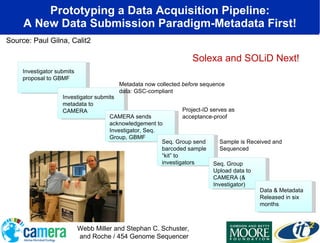
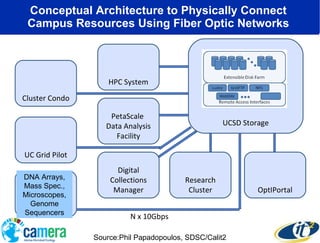

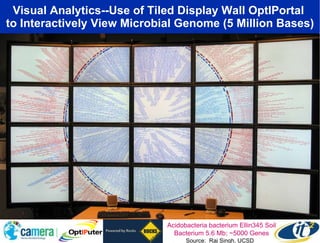





The document discusses the advancements in metagenomics, emphasizing the importance of microbial life in ecosystems and the limitations of traditional research methods. It introduces the CAMERA project, a cyberinfrastructure aimed at handling vast amounts of metagenomic data while facilitating user collaboration and access across a global community. Additionally, it outlines future trends in metagenomic data generation, standardization, and the integration of cloud computing.








![[2013.12.02] Mads Albertsen: Extracting Genomes from Metagenomes](https://cdn.slidesharecdn.com/ss_thumbnails/2013-131202013655-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)






![[2013.09.27] extracting genomes from metagenomes](https://cdn.slidesharecdn.com/ss_thumbnails/2013-130927034103-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)








![[2013.10.29] albertsen genomics metagenomics](https://cdn.slidesharecdn.com/ss_thumbnails/2013-131029070115-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)


![[13.07.07] albertsen mewe13 metagenomics](https://cdn.slidesharecdn.com/ss_thumbnails/13-07-07albertsenmewe13metagenomics-130707072221-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)



























































