Downloaded 419 times











![FIRST-ORDER ABSORPTION MODEL
Ka KE
Drug Blood Excretion
first order
From equ. dX = dXev - dxe
dt dt dt
Differentiating above equ. We get,
dX = Ka Xa – KEX, Ka= absorption rate const.
dt Xa= amt of drug remaining
to be absorbed.
Integrating above equ.,
X =
[ K T K t ]
K FX - - -
( - )
a o e E e a
K K
a E
13](https://image.slidesharecdn.com/aseminarononetwocompartmentopenmodelextravascularadministration-140930002043-phpapp02/75/A-seminar-on-one-two-compartment-open-model-extra-vascular-administration-13-2048.jpg)






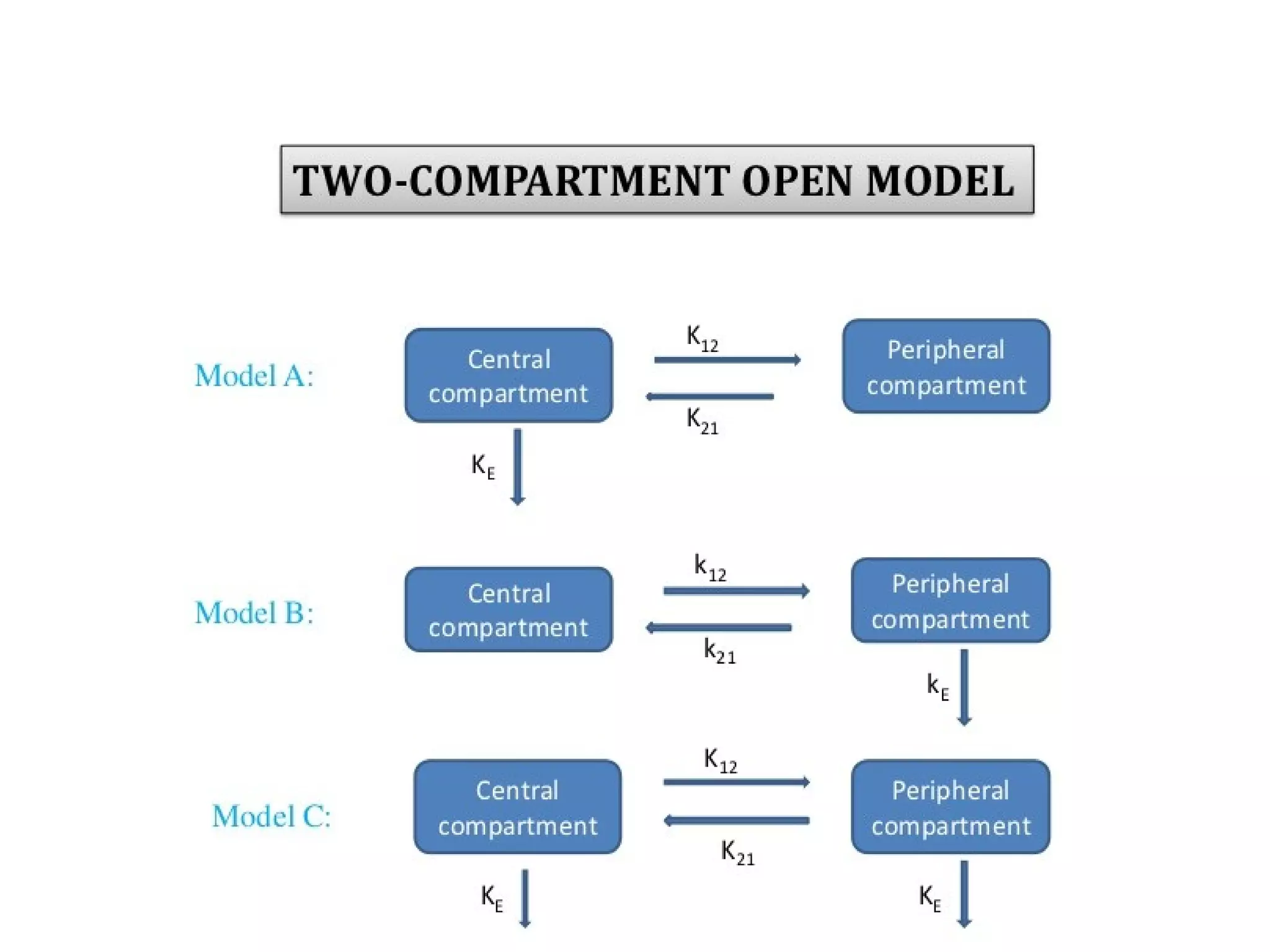







This document summarizes one and two compartment open models for extravascular drug administration. It describes how compartment models are used to simplify drug distribution and elimination processes in the body. A one compartment open model is presented, showing drug absorption from extravascular administration followed by distribution and elimination from the body compartment. Equations are provided to describe drug behavior under zero-order and first-order absorption. Methods for estimating the absorption rate constant like residuals and Wagner-Nelson are also summarized. Finally, a two compartment open model is briefly introduced.



































































