








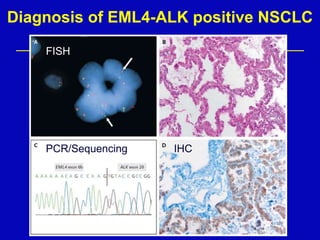
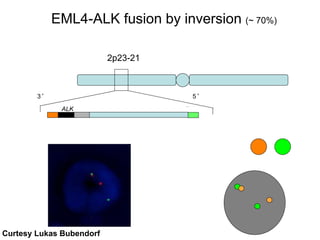






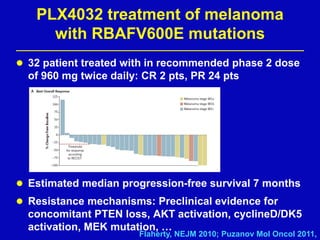










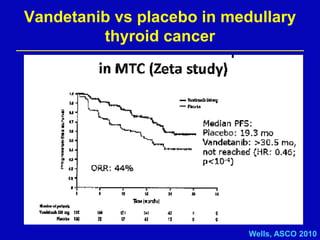




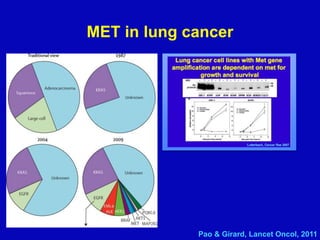

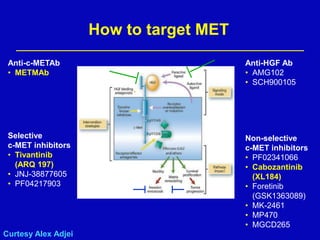



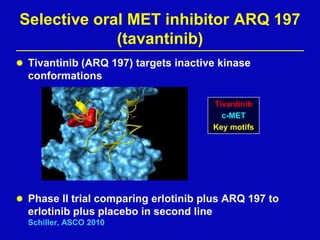




Rolf Stahel presented a document on various oncogenic pathways and targeted therapies. The document discussed: 1) The ALK pathway and its role in cancers like NSCLC and neuroblastoma. Drugs like Crizotinib have shown responses in ALK-positive NSCLC. 2) The RET pathway's role in medullary thyroid cancer. Drugs like Vandetanib have shown responses in RET-mutated MTC. 3) The Hedgehog pathway's role in basal cell carcinoma and medulloblastoma. Inhibitors like GDC-0449 have induced responses in Hedgehog pathway-driven tumors.









































































