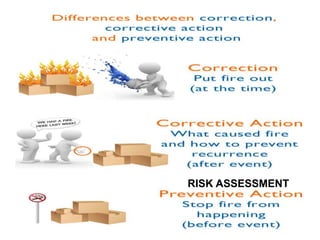
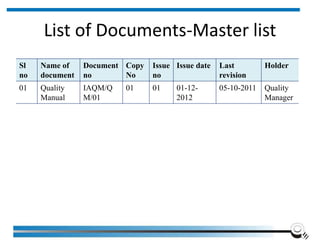
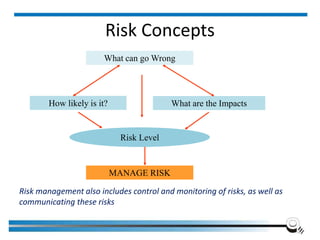
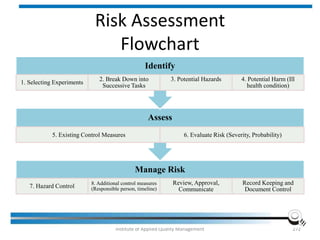
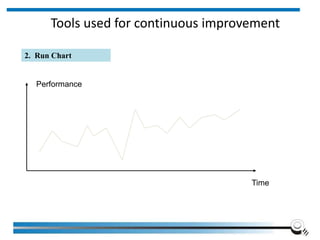
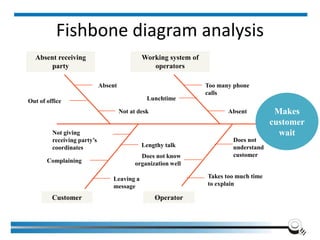
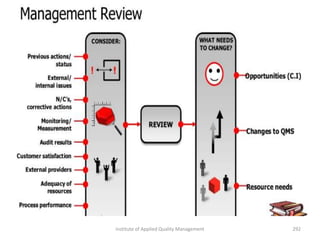
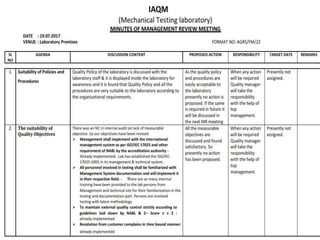
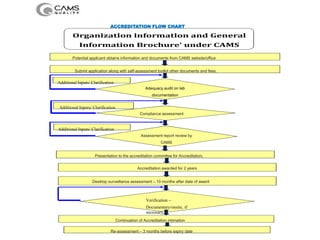
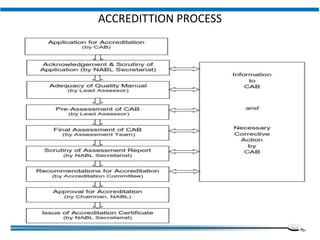
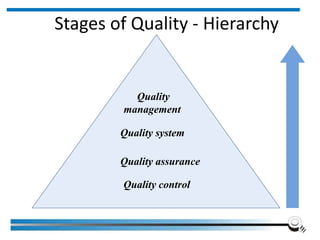
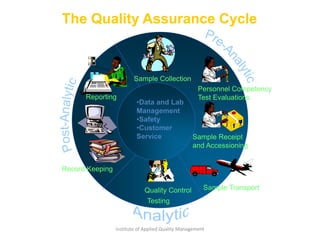
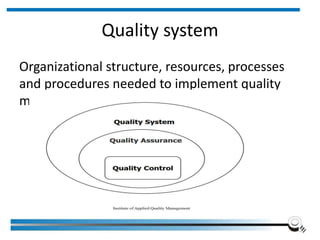
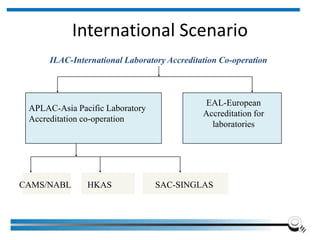
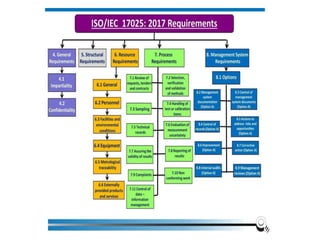
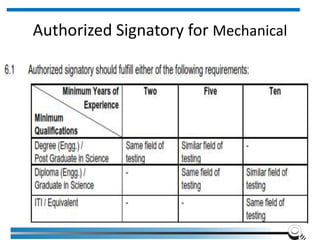
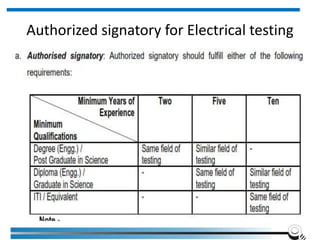
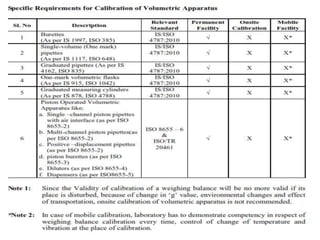
The document discusses the ISO/IEC 17025:2017 requirements related to the competence and quality management of laboratories, emphasizing the importance of impartiality, confidentiality, and precise operation. It outlines the structural, resource, and management system requirements necessary to ensure quality assurance and control, as well as the significance of laboratory accreditation for demonstrating technical competency. Key elements include competency assessments, risk identification, and the need for suitable facilities and equipment to support laboratory activities.



































































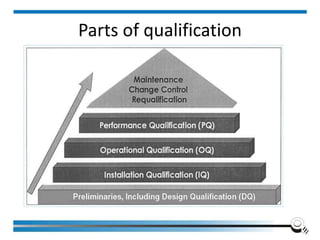
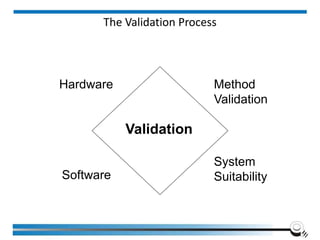
![Equipment Validation Overview
[To establish] documented evidence which provides a high
degree of assurance that a specific process will consistently
produce a product meeting pre-determined specifications and
quality attributes.
Institute of Applied Quality Management 78](https://image.slidesharecdn.com/5-240713063929-59a409d9/85/17025-2017-Training-Manual-for-the-Laboratory-78-320.jpg)




















































































































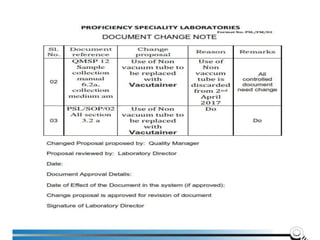
![7.8.8.1 When an issued report needs to be changed,
amended or re-issued, any change of information
shall be clearly identified and, where appropriate, the
reason for the change included in the report.
7.8.8.2 Amendments to a report after issue shall be
made only in the form of a further document, or data
transfer, which includes the statement “Amendment to
Report, serial number... [or as otherwise identified]”, or
an equivalent form of wording.
7 Process requirements](https://image.slidesharecdn.com/5-240713063929-59a409d9/85/17025-2017-Training-Manual-for-the-Laboratory-203-320.jpg)