Downloaded 757 times














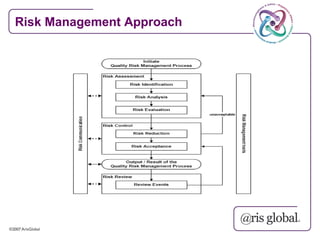








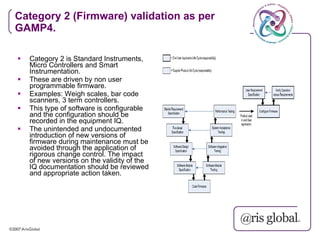


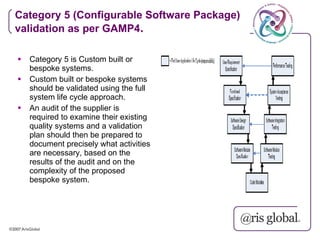




This document provides an overview of GAMP (Good Automated Manufacturing Practice) guidelines for validation of computer systems used in regulated industries. It discusses the history of GAMP, key terms and concepts in validation like validation life cycle, risk management, categories of software. It also summarizes the validation requirements for different categories of software and records as per GAMP-4 guidelines. The document emphasizes that validation is important to ensure computer systems consistently produce intended results and meet safety standards.















































