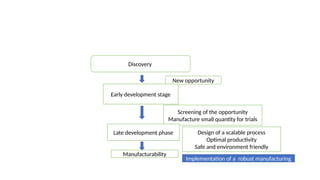
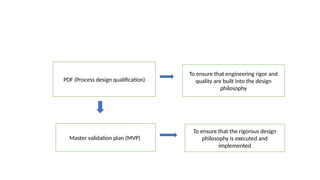
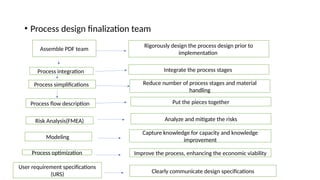
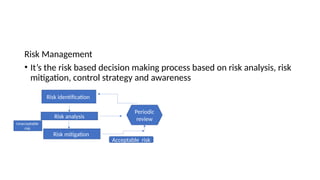
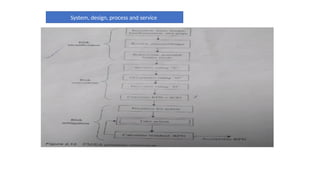
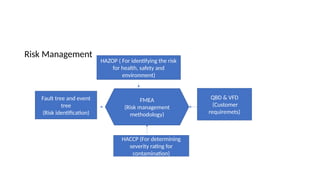
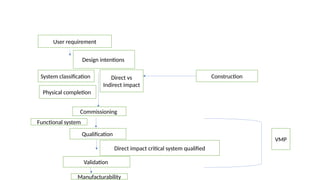
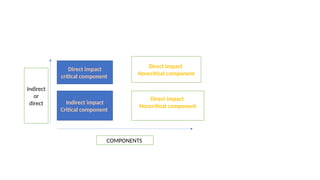
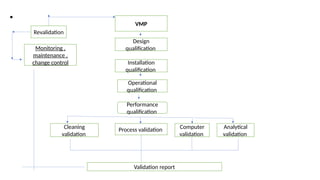
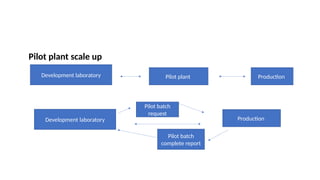
The document outlines guidelines for pharmaceutical development and validation processes as per ICH directives, emphasizing the integration of quality risk management and robust manufacturing practices. It details specific stages of process validation, including design, operational, and performance qualifications, along with various strategies for ensuring compliance with quality standards. The document also discusses critical process parameters, user requirements, and the significance of effective risk analysis in maintaining product quality and safety.

































































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)











