Downloaded 1,309 times


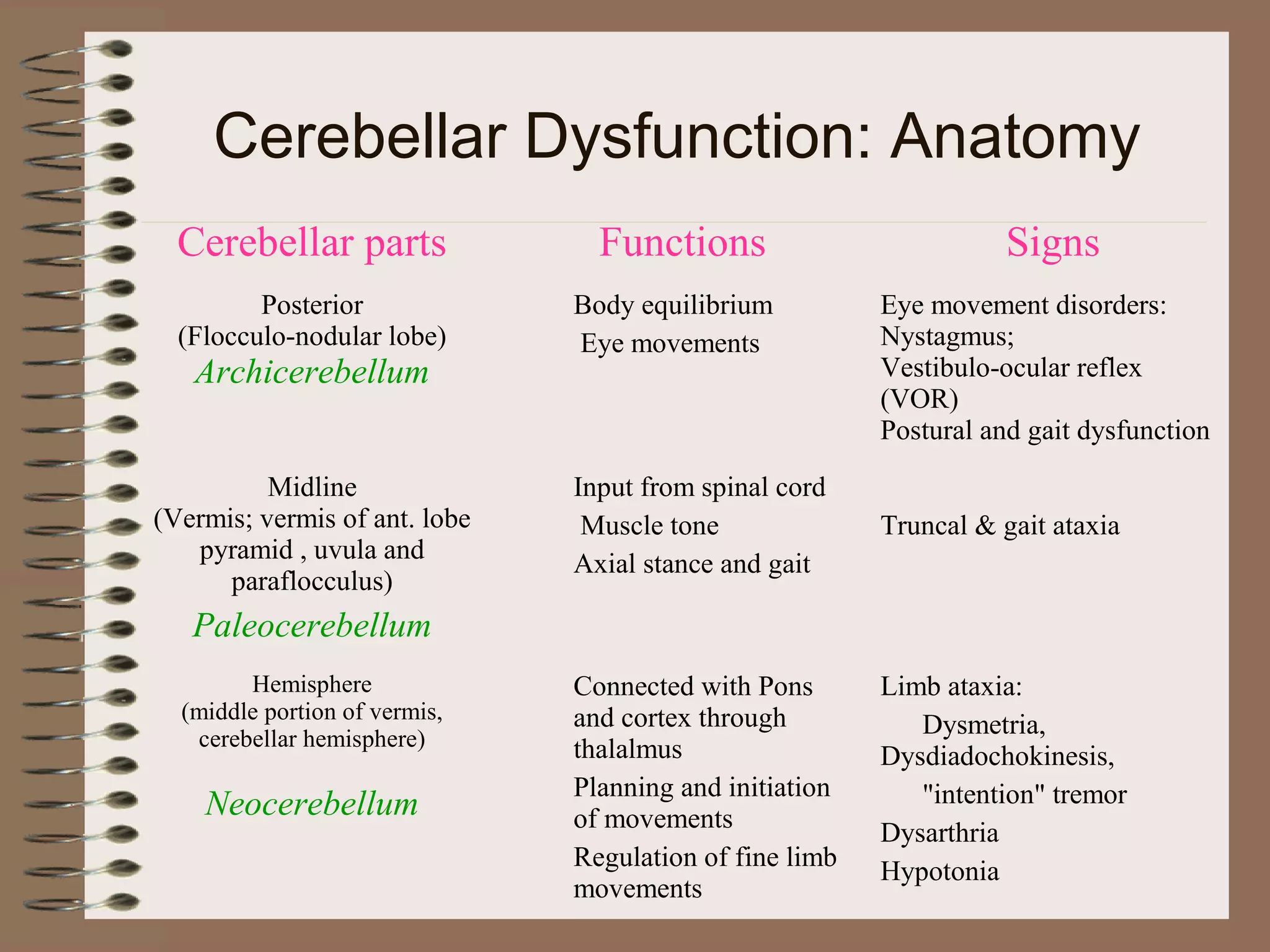








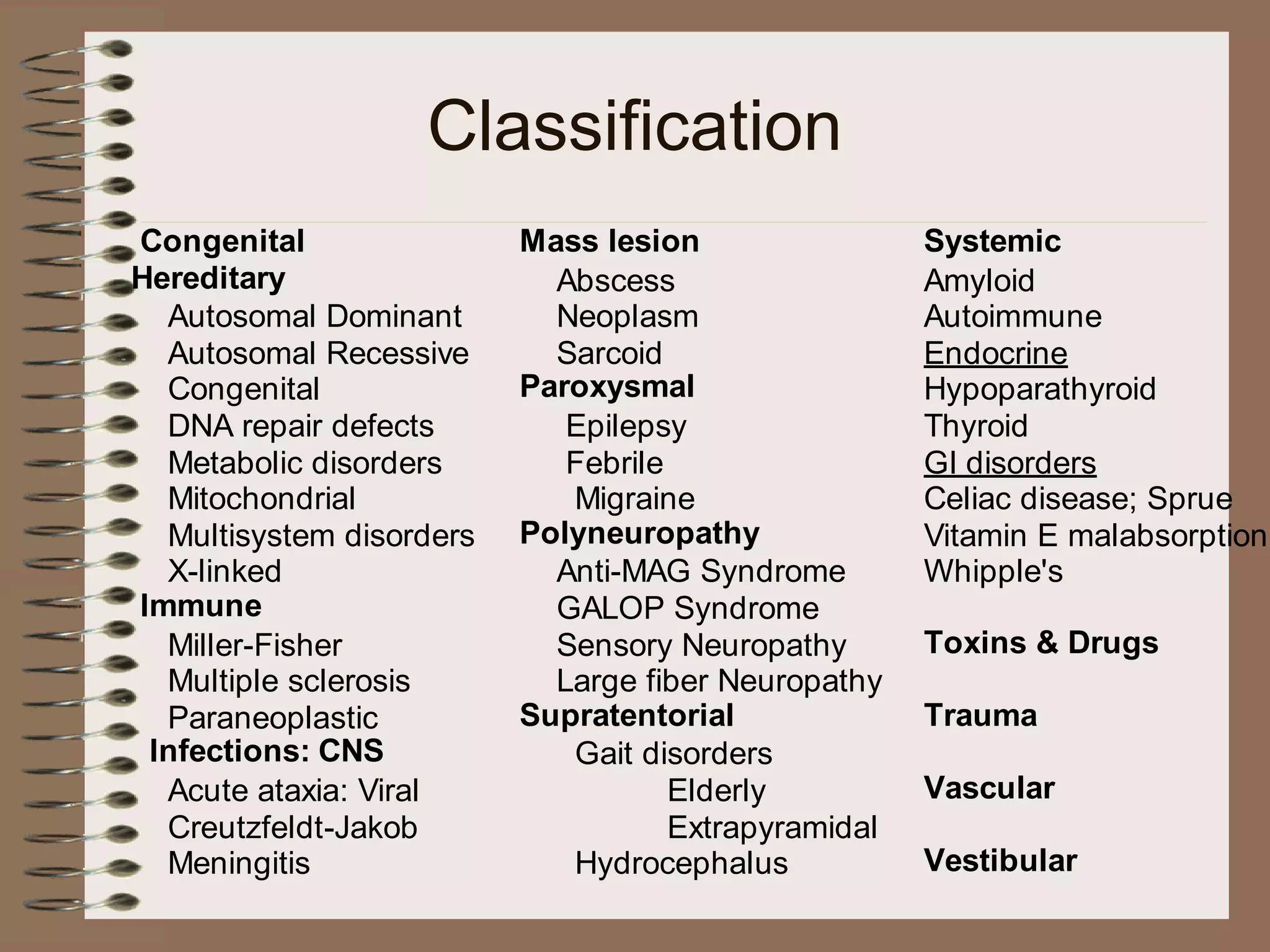












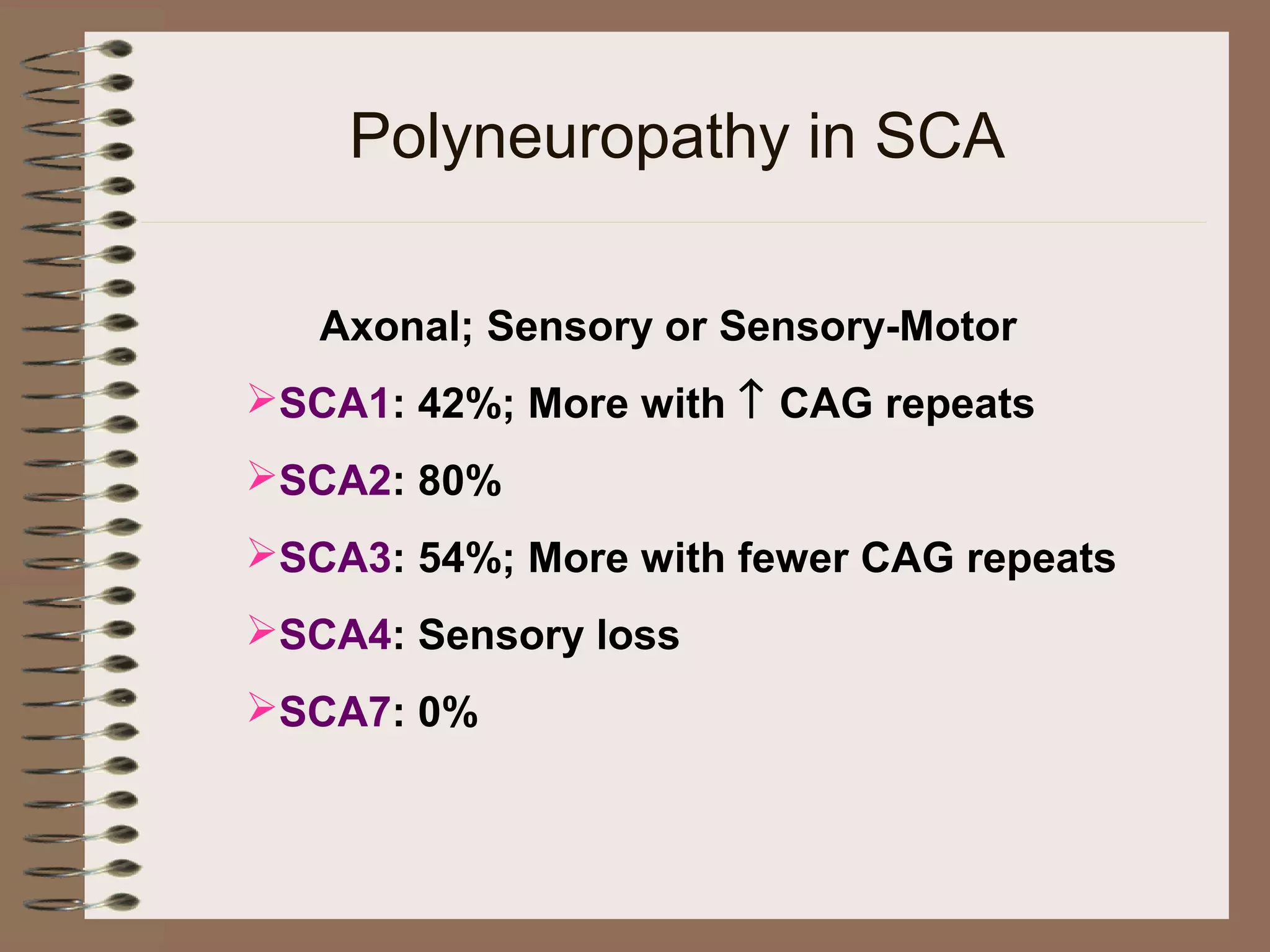


















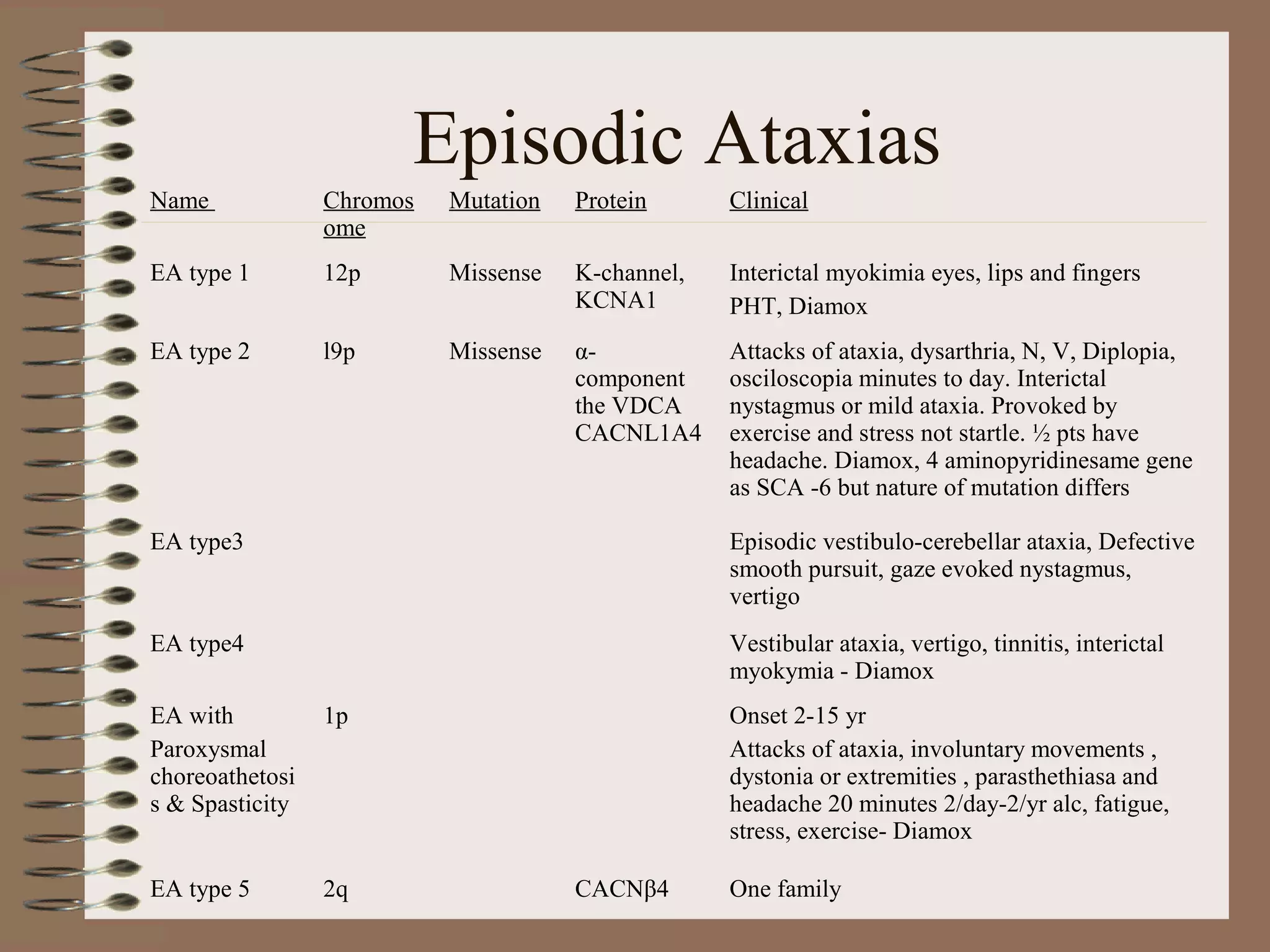









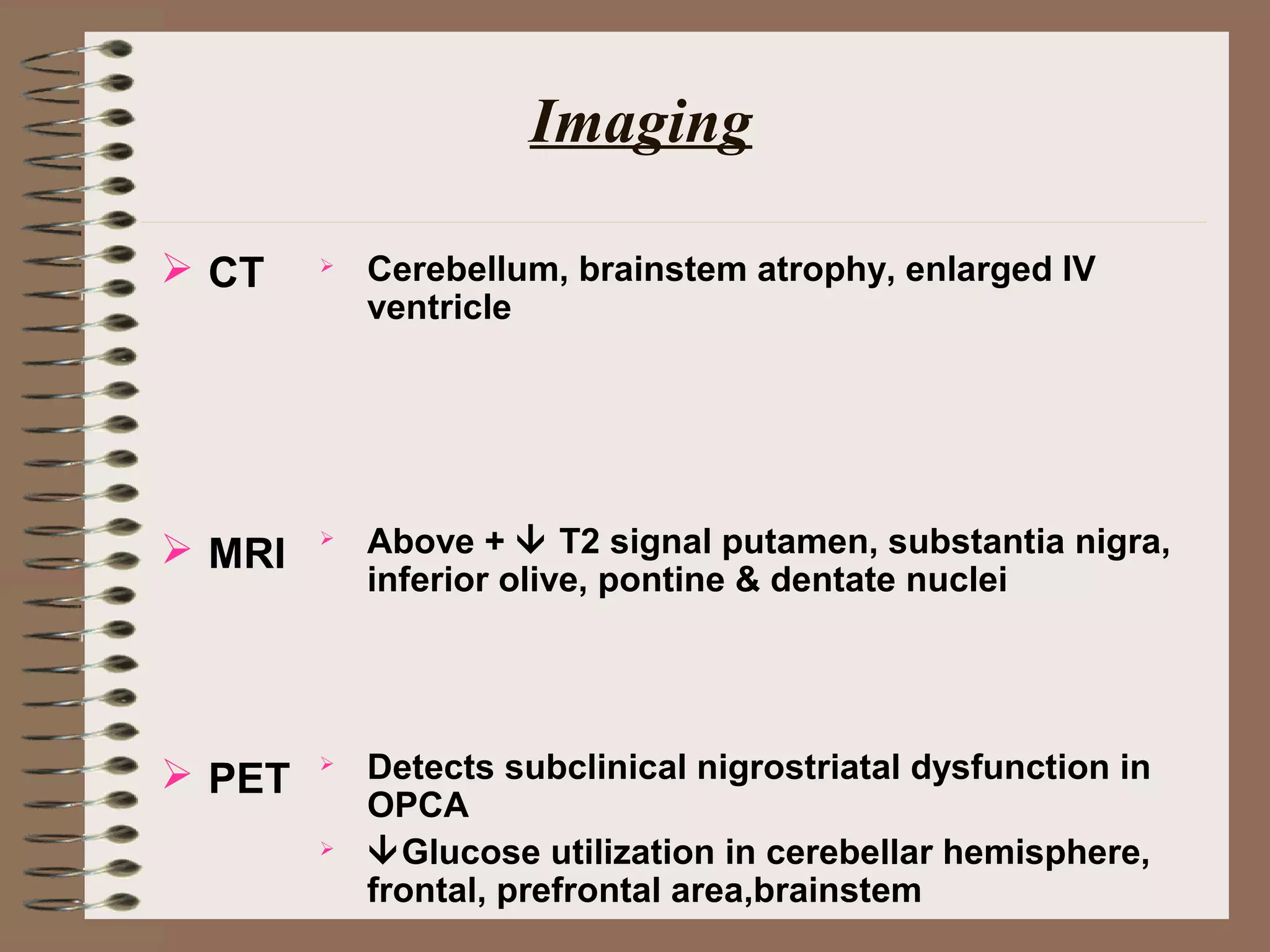




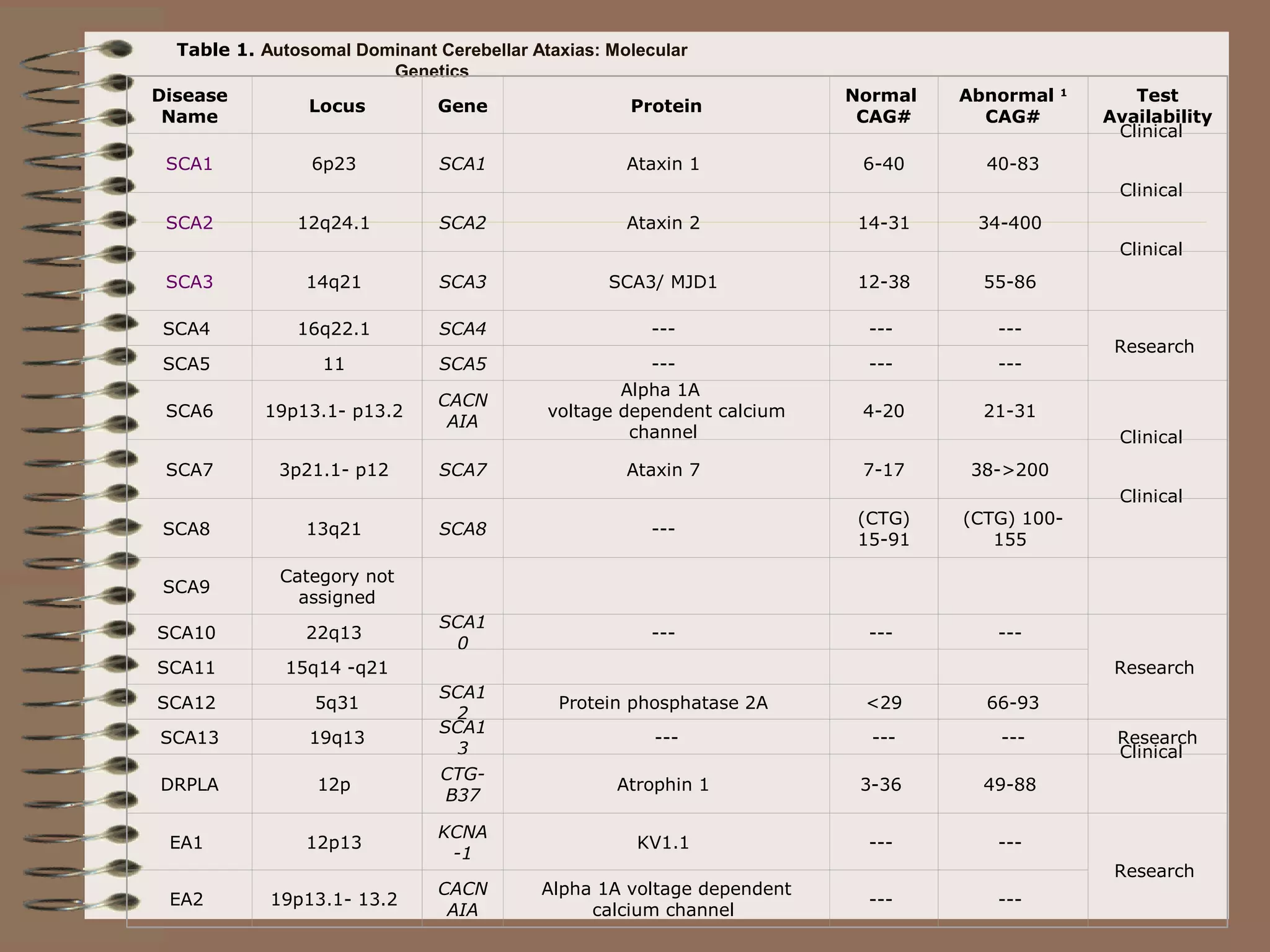
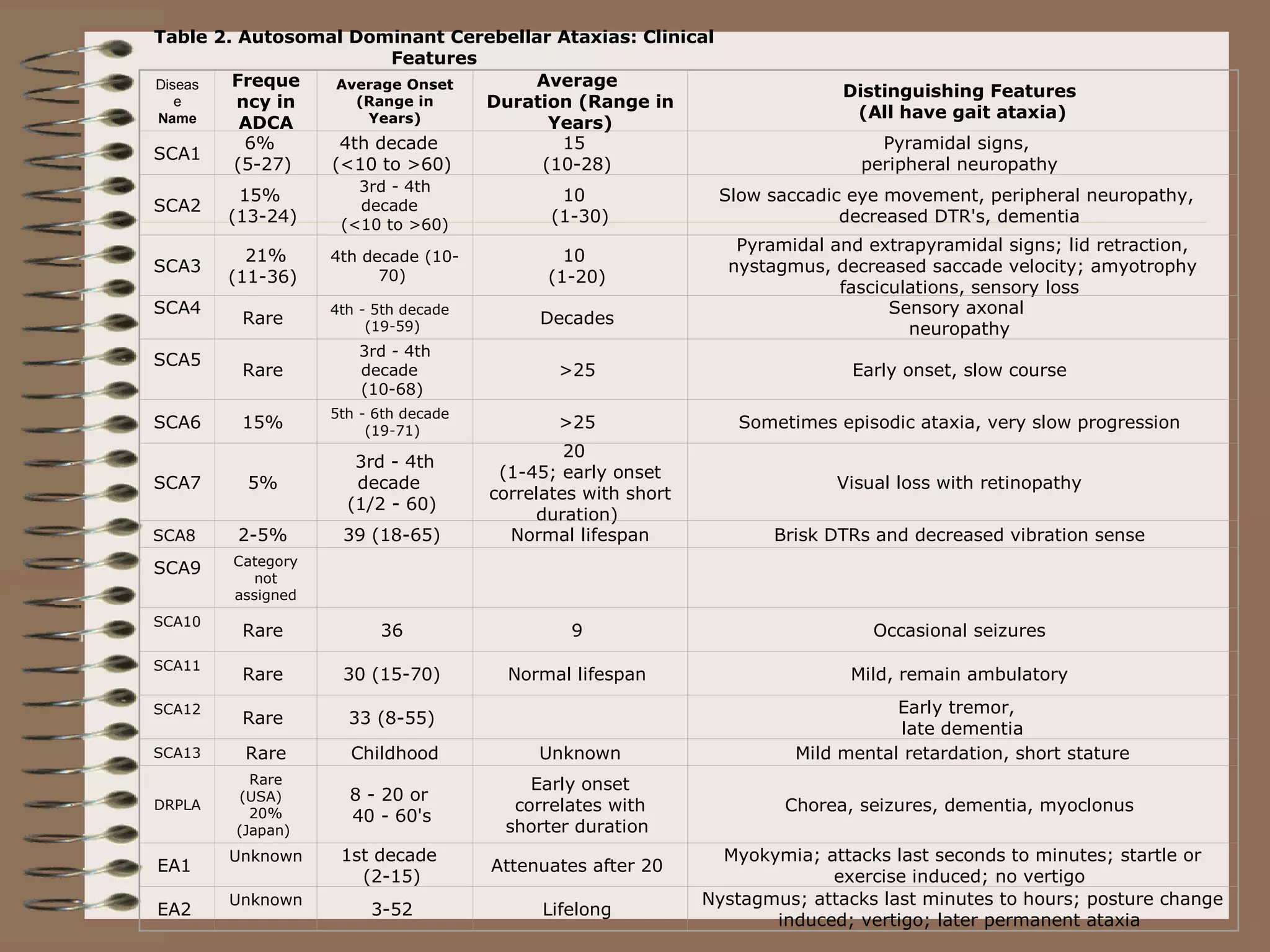
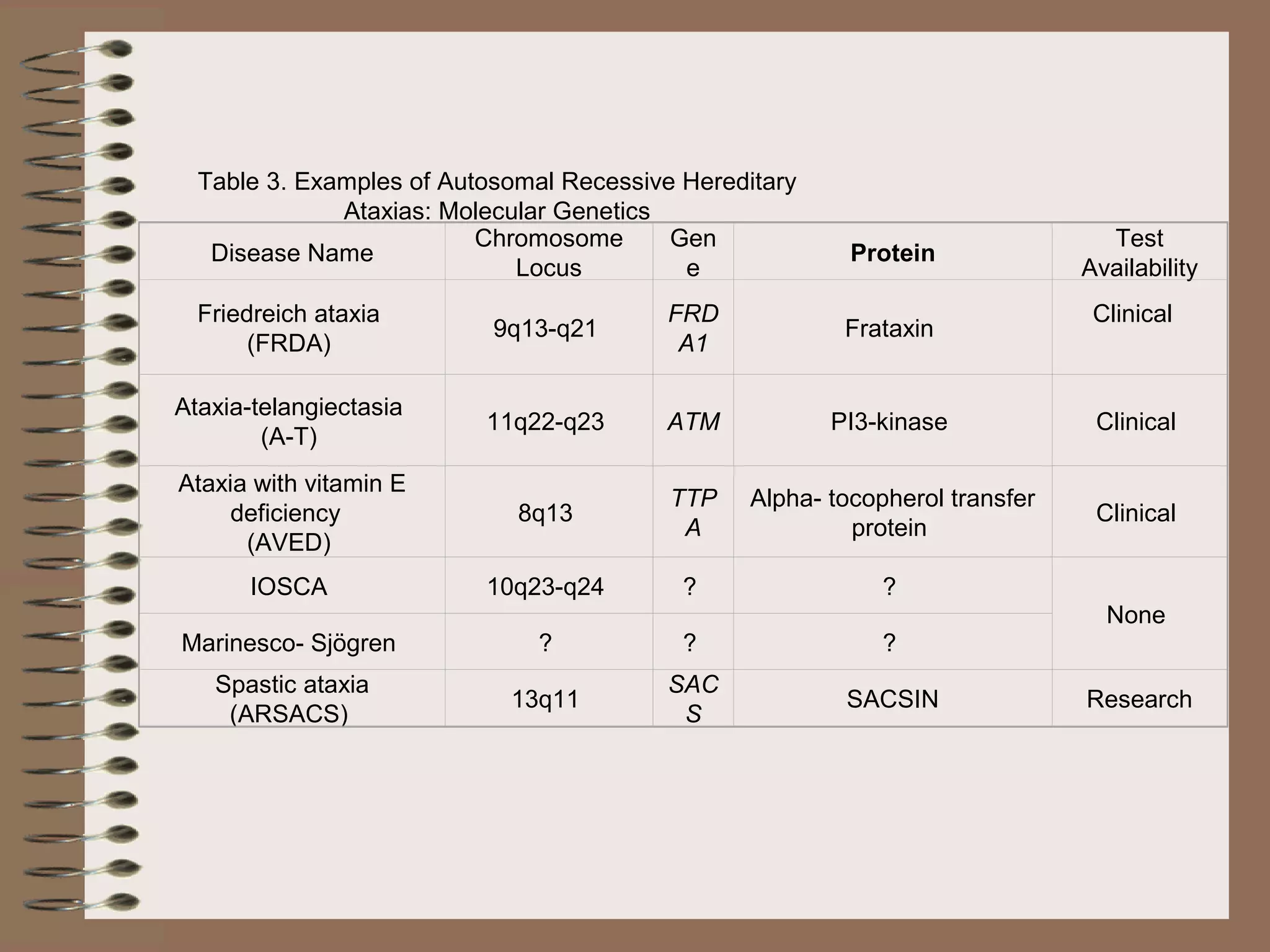
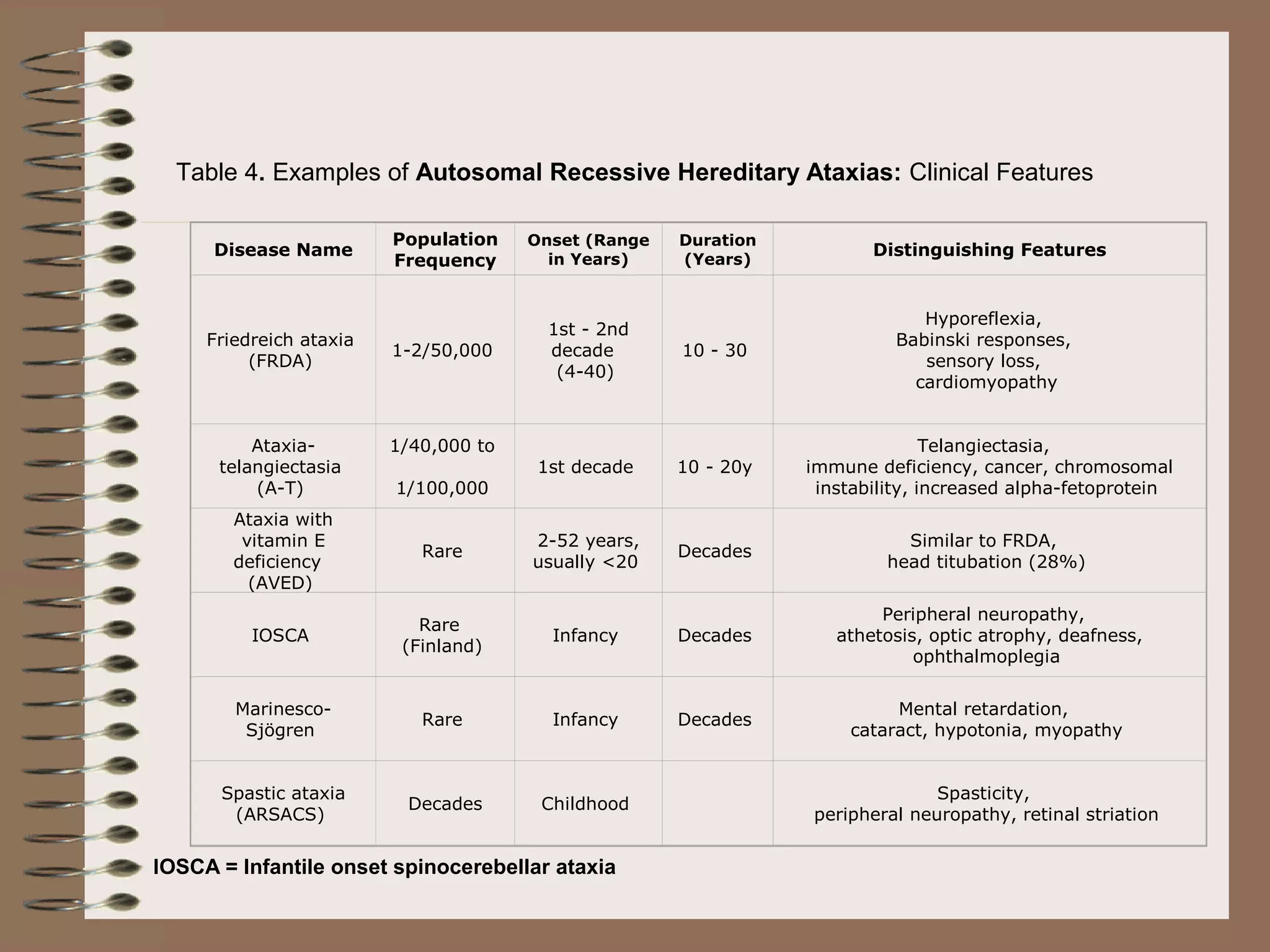



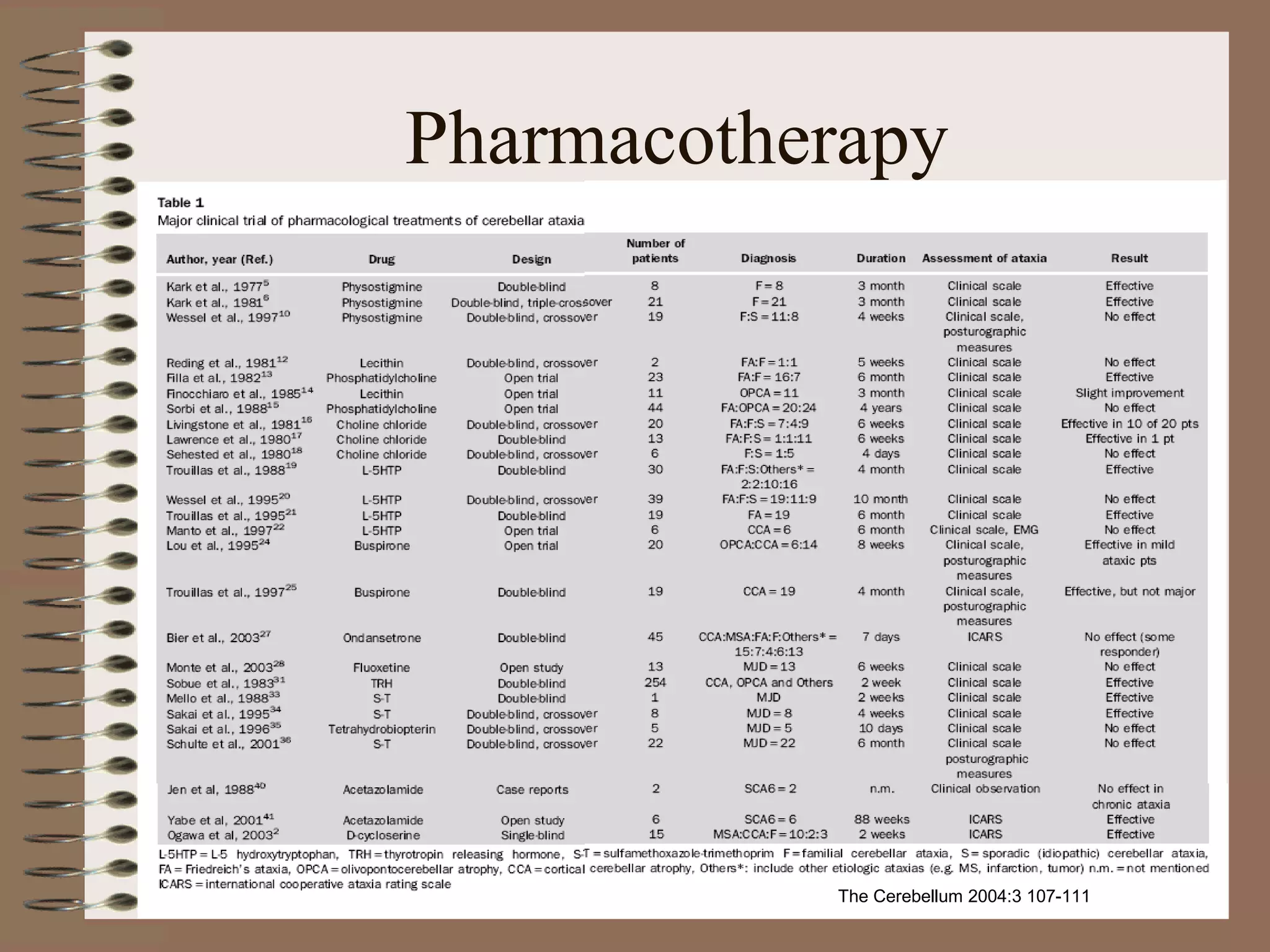
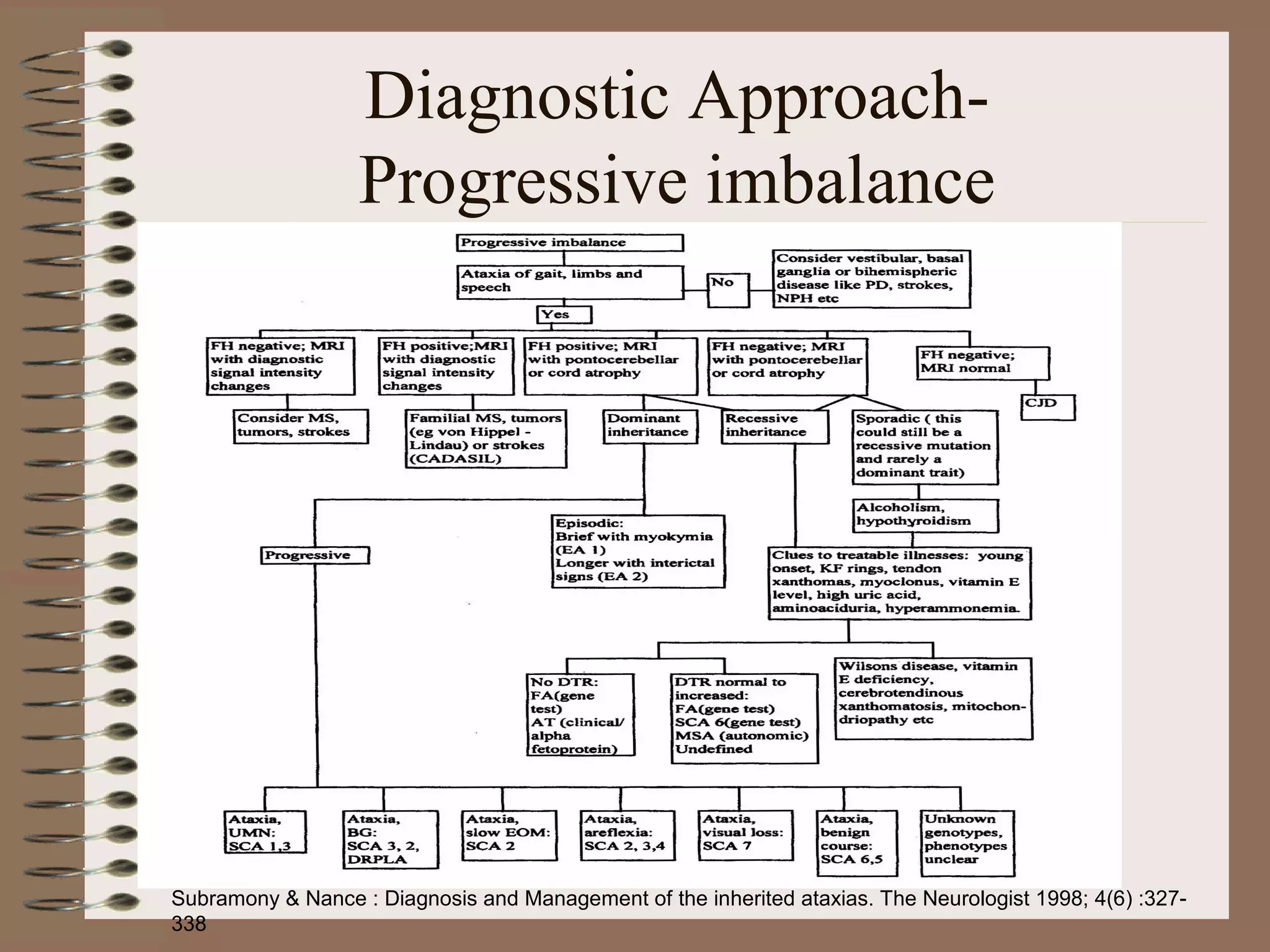
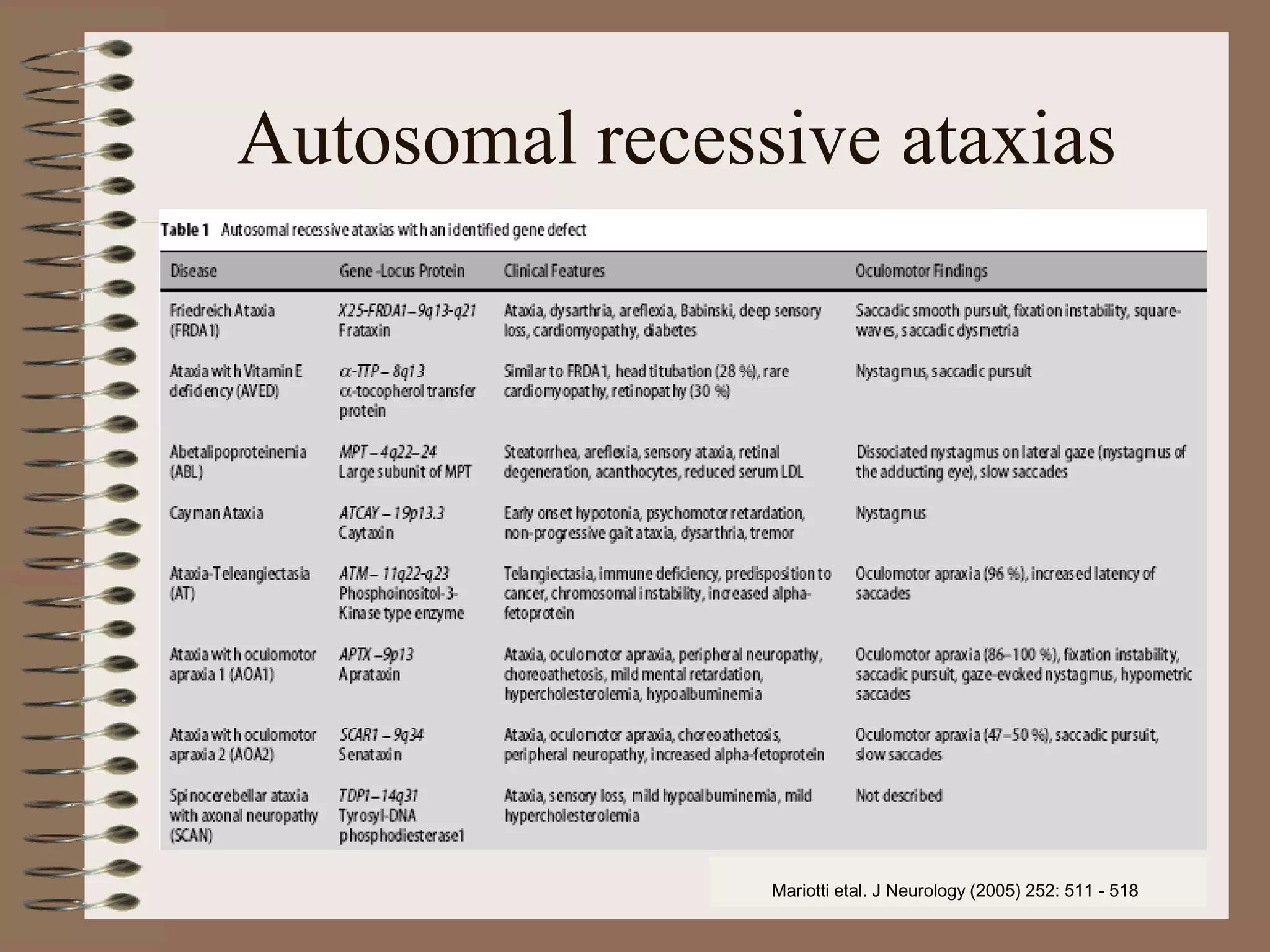
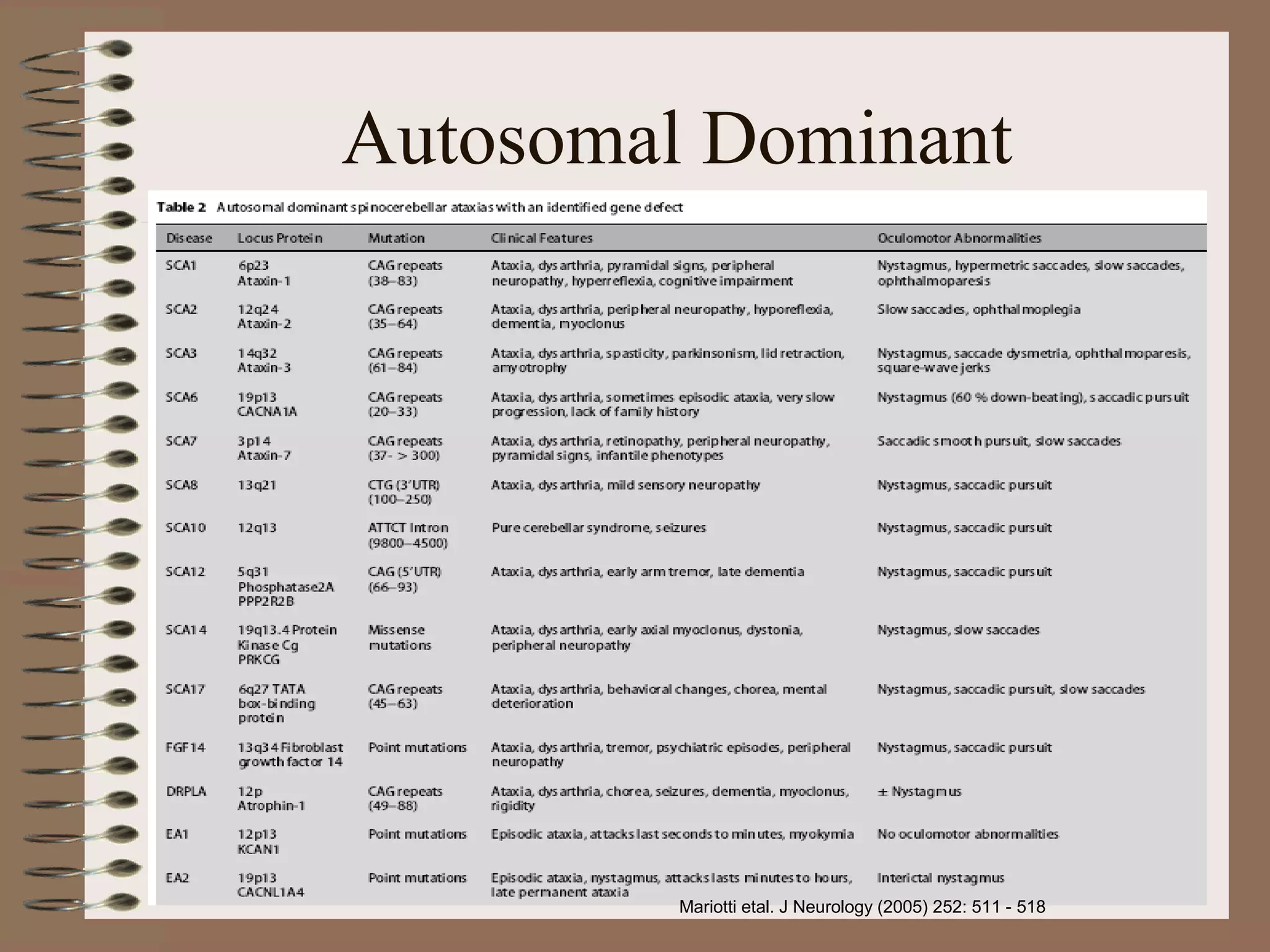


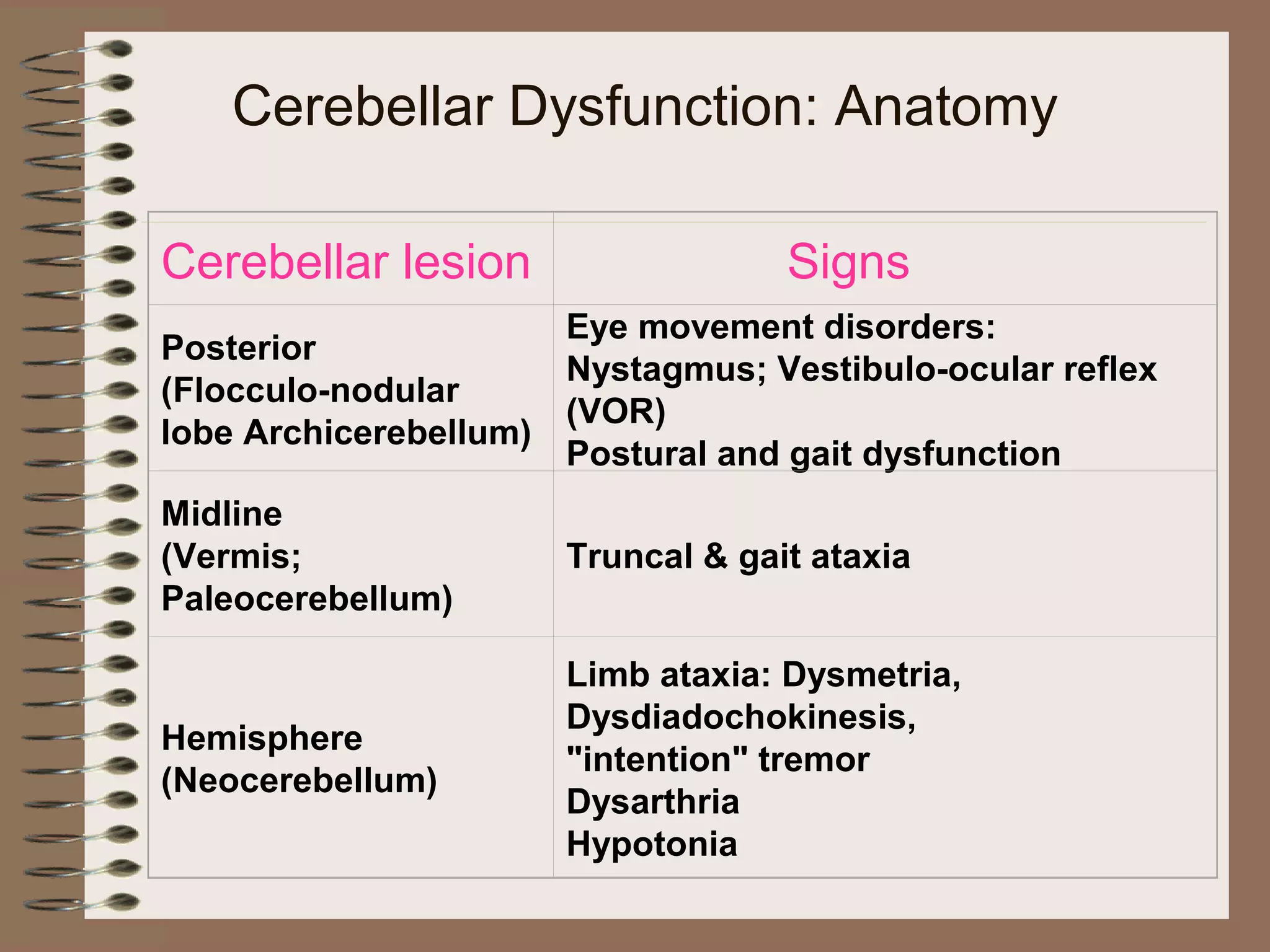


This document provides an overview of ataxia, including its definition, causes, clinical features, classifications, hereditary forms, and spinocerebellar ataxia (SCA). Key points include: Ataxia is caused by dysfunction of the cerebellum and its pathways, resulting in loss of coordination. Common clinical features are gait and limb ataxia, dysarthria, and gaze abnormalities. Causes include genetic, paraneoplastic, infectious, autoimmune, and others. Hereditary forms include autosomal dominant and recessive SCAs, Friedreich's ataxia, and more. SCA is the most common hereditary ataxia and subtypes are distinguished by their clinical



































































