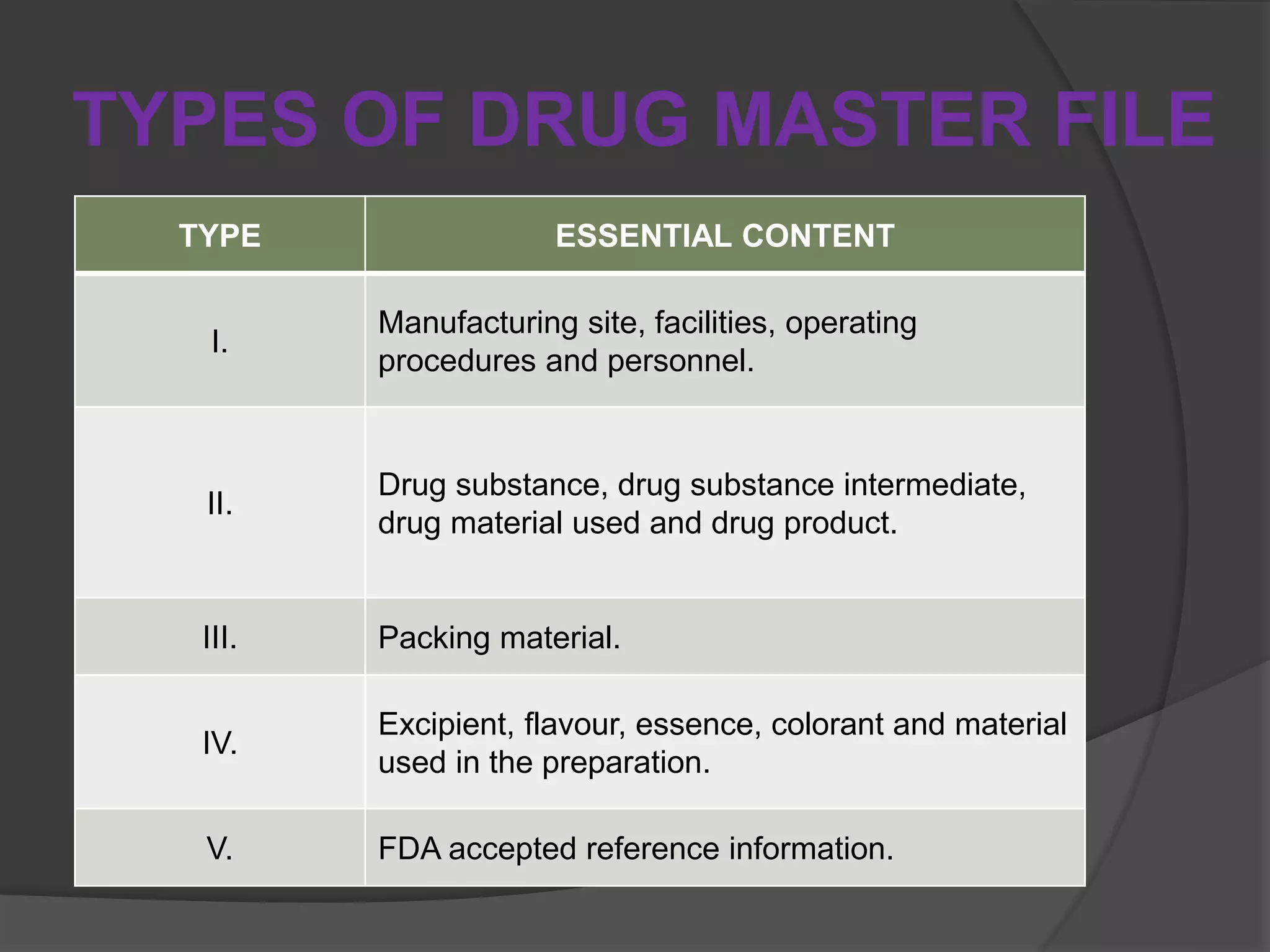
A Drug Master File (DMF) is a confidential submission to the FDA containing detailed information about the manufacturing and quality control of human drugs, though it is not mandatory. DMFs support applications like IND, NDA, and ANDA, and help establish the credibility of API manufacturers. The submission includes various types covering manufacturing sites, drug substances, packaging, excipients, and requires an annual update to confirm the information remains current.
































































![Drug master file ppt [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/drugmasterfilepptautosaved-200130192621-thumbnail.jpg?width=640&height=640&fit=bounds)























