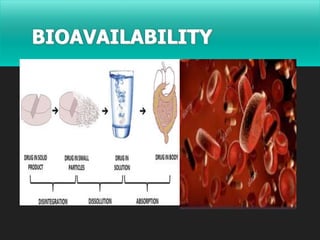
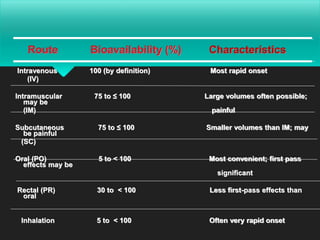
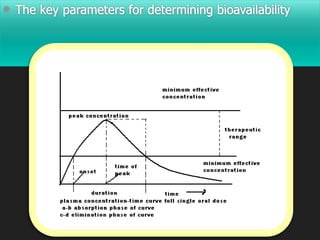
Bioavailability is defined as the rate and extent to which an active drug reaches systemic circulation. Objectives of bioavailability studies include developing new formulations and determining the effects of excipients, patient factors, and drug interactions on absorption. Bioavailability can be assessed using pharmacokinetic methods like determining blood concentrations over time to calculate AUC, Cmax, and Tmax, or by measuring urinary drug excretion. Bioequivalence studies establish equivalence between test and reference drug products using randomized crossover or parallel designs in accordance with regulatory guidelines. Key parameters compared include AUC, Cmax, and the time to reach Cmax to ensure bioequivalent drug products have similar rates and extents of absorption.














































![Rate limiting steps in drug absorption [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/ratelimitingstepsindrugabsorptionautosaved-200623174235-thumbnail.jpg?width=640&height=640&fit=bounds)















































