Downloaded 18 times




















![Rate limiting steps in drug absorption [autosaved]](https://image.slidesharecdn.com/ratelimitingstepsindrugabsorptionautosaved-200623174235/85/Rate-limiting-steps-in-drug-absorption-autosaved-23-320.jpg)

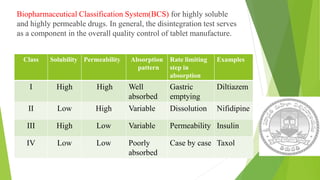
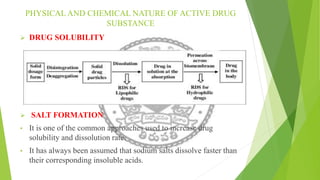
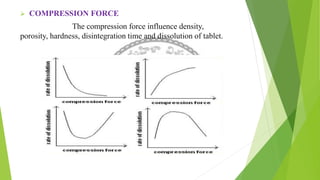
The document discusses rate limiting steps in drug absorption, focusing on the movement of unchanged drugs from administration sites to plasma and the factors influencing this process, including disintegration, dissolution, and absorption rates. Key factors affecting drug absorption include drug solubility, properties of excipients, and manufacturing methods. The conclusion emphasizes the importance of understanding these processes to determine the rate and extent of drug absorption.














































































