Downloaded 98 times






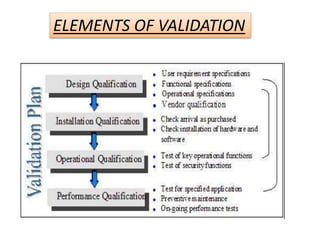
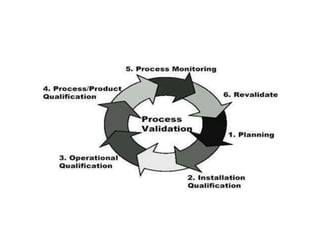






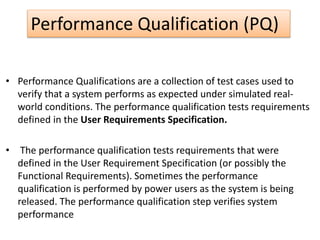


















This document provides an overview of validation in the pharmaceutical industry. It discusses the concepts, types, elements and phases of validation including design qualification, installation qualification, operational qualification, performance qualification and process validation. Validation is required to ensure manufacturing processes are capable of consistently producing quality products and involves establishing documented evidence through qualification and monitoring activities. The document outlines the key principles and guidelines for validation as well as the roles and responsibilities of those involved.
















































































