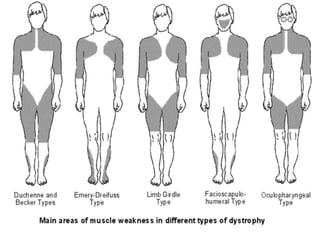
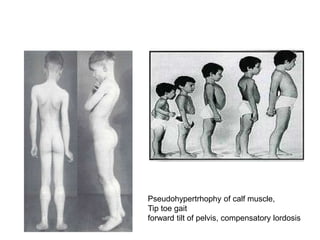
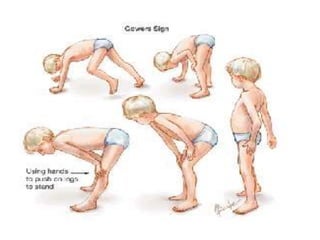
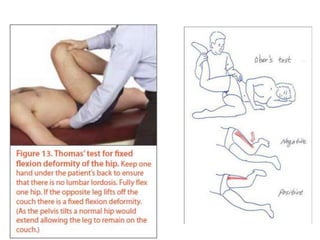
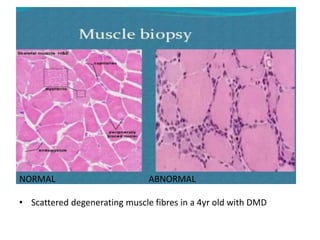
Muscular dystrophy is a genetic disease that causes the muscles to weaken over time. There are several types but Duchenne muscular dystrophy is the most common and severe, affecting boys. It is caused by an absence of dystrophin protein which leads to muscle cell damage. Symptoms start in early childhood and include difficulty walking, joint contractures, and loss of ambulation in the teen years. Management is multidisciplinary and focuses on maintaining mobility and function as long as possible, treating complications, and palliative care as the condition progresses. Life expectancy for Duchenne patients is usually early 20s.