Vincent Timmerman - 'Neuropatías periféricas hereditarias'
•
4 likes•1,479 views

1. The document summarizes research on genetic causes and pathogenic mechanisms underlying various forms of Charcot-Marie-Tooth (CMT) disease and distal hereditary motor neuropathies (dHMN). It describes mutations found in genes involved in protein folding, axonal transport, cytoskeletal stability and other pathways. 2. Research shows that mutations in small heat shock proteins HSPB1 and HSPB8, which are implicated in CMT and dHMN, cause motor neuron dysfunction and protein aggregation. Studies using cell and animal models demonstrate disease-relevant gain of toxic function from these mutations. 3. The small heat shock proteins HSPB1 and HSPB8 are normally involved in

Recommended
Recommended
More Related Content
What's hot
What's hot (19)
Similar to Vincent Timmerman - 'Neuropatías periféricas hereditarias'
Similar to Vincent Timmerman - 'Neuropatías periféricas hereditarias' (20)
More from Fundación Ramón Areces
More from Fundación Ramón Areces (20)
Recently uploaded
Recently uploaded (20)
Vincent Timmerman - 'Neuropatías periféricas hereditarias'
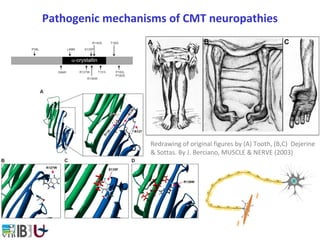
- 1. Pathogenic mechanisms of CMT neuropathies Redrawing of original figures by (A) Tooth, (B,C) Dejerine & Sottas. By J. Berciano, MUSCLE & NERVE (2003)
- 2. Exploring the genetic causes of PNS degeneration Timmerman, Strickland, Züchner GENES (2014) + updates AIFM1, COX6A1, VCP, …
- 3. CMT1B DSS, CH, CMT2 CMT2B AR dHMN DSMA+ CMT2D CMT4A ARCMT2 CH, CMT1, DSS HMSN-R CMT1A HNPP DSS HNA CMT1C CMT4B1 1 2 3 5 8 X 12 17 9 DSMA ALS4/dHMN CMTX1 11 CMTX3 dHMN V dHMN VIIa dHMN VIIb HMSN-L DSS HMSN- P 10 dHMN II-CMT2L ARCMT2 CMT2C + Con.distal SMA HSN2 dHMN VI dHMN V’ MPZ LMNA EGR2 ARHGEF10 CTDP1 PRPS1 19 CCFDN HSN III ARCMT2A 1815 AS NDRG1 GDAP1 MTMR2 BSCL2 IGHMBP2 SLC12A6 SBF2 dHMN-J DI-CMTA DI-CMTB NEFL GARS SH3TC2 HINT1 HARS ARCMT2B CMT4F/ DSS NGFB HSAN IV CMT4B2 BICD2 PRX GAN HSPB8 COX6A1 LITAF/ SIMPLE 16 CMT2F CMT2EYARS RAB7 DCTN1 SETX 22 CMT1 +PM +WHS CMT4B3 SOX10 SBF1 reduced NCV DI-CMTC HSNI + cough GER PMP22 HSPB1 IKBKAP LRSAM1 FGD4 GAN CMT2G HSAN V NTRK1 DNM2 PLEKHG5 SEPT9 CMT4H dHMN X CMTX5 WNK1 CMT4JFIG4 5 6 GJB1 CMTX2/X4 MFN2/KIF1B CMT2A MED25 TRPV4 ATP7A HSNIIb CCT5 HSAN IATL1 SPTLC2 14 KARS DI-CMT AARS CMT2 Hk1 FBLN5 INF2 DYNC1H1 CMT+CL CMT+KD CMT2O REEP1 dHMN V MT-ATP6A CMT2DNAJB2 AR dHMN dHMN IicHSPB3 CMTX6PDK3 20 HMN atypicalVAPB CPEOM3TUBB3 HSANI-EDNMT1 KIF1A HSAN II-C TFG HSAN+ SPG FAM134B HSAN VIDST SCN9A HSAN II-D SLC5A7 HSN ISPTLC1 7 HMSN+ataxiaIFRD1 4 AR- CMT2 TRIM2 GJB3 Sens PN + Hearing loss DI-CMTGNB4 CMT2QDHTKD1 HSANSCN11A SURF1 CMT4 CMT4C ARCMT +NM PN sens FBXO38 AIFM1 VCP CMT2? Timmerman, Strickland, Züchner GENES (2014) + updates Baets, De Jonghe, Timmerman Curr Opin Neurol (2014)
- 4. Overlapping molecular pathological themes link CMT and hereditary spastic paraplegias Timmerman, Clowes and Reid, Experimental Neurology (2013) + updates ATL3 VCP
- 5. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders Novarino et al. Science (2014)
- 6. HMSN hereditary motor and sensory neuropathy = ‘CMT’ Motor Sensory & Autonomic HSAN hereditary sensory & autonomic neuropathy HMN hereditary motor neuropathy Harding & Thomas, J. Med. Genet (1980), J. Neurol. Sci (1980) and Brain (1980) Harding, A.E. (1993) In: Dyck, Thomas, Griffin, Low and Poduslo (Eds) In: Peripheral Neuropathy Dyck, P.J. (2005) In: Peripheral Neuropathy Various kinds of inherited peripheral neuropathies
- 7. SUBTYPE INH GENE PHENOTYPE MUTATIONS dHMN1 AD 7q34-36 typical HMN (juvenile onset) - dHMN2A AD HSPB8 typical HMN (adult onset) 4 dHMN2B AD/AR HSPB1 typical HMN (juvenile and adult onset) 18 dHMN2C AD HSPB3 typical HMN 1 dHMN2D AD FBXO38 distal SMA with predominance weakness of the calf muscles 1 DSMA4 AR PLEKHG5 proximal and distal muscle atrophy, scoliosis, respiratory failure 3 dHMN AD SETX HMN with pyramidal signs (juvenile ALS4) 4 DSMA5 AR DNAJB2 typical HMN 1 dHMN5A AD GARS predominant weakness in hands 10 dHMN5B AD REEP1 predominant weakness in hands, pyramidal signs 1 dHMN5C AD BSCL2 predominant weakness in hand, Silver syndrome, rarely sensory symptoms (CMT2D) 2 dHMN6 AR IGHMBP2 juvenile onset; SMARD, diaphragmatic SMA 55 dHMN7A AD SLC5A7 typical HMN; vocal cord paralysis 1 dHMN7B AD DCTN1 bulbar and facial weakness 1 dHMNJ AR 9p21.1-12 juvenile onset; pyramidal signs - SMARD2 X-linked LAS1L distal muscle weakness, respiratory failure, diaphragm paralysis 1 SMAX3 X-linked ATP7A typical HMN (X-linked distal SMA) 2 SMALED AD BICD2 congenital AD-SMA 4 DYNC1H1 congenital; contractures; predominant in lower limbs; pyramidal signs; learning difficulties; cortical migration defects 6 PNMHH AD MYH14 typical HMN; distal myopathy; hoarseness; hearing loss 1 SPSMA AD TRPV4 scapuloperoneal SMA with vocal cord paralysis 6 dHMN AD AARS typical HMN 1 ARAN-NM AR HINT1 distal muscle weakness; neuromyotonia involving the hands 8 134 mutations in 21 genes (updated July 2014)
- 8. Muscle Motor neuron High metabolism and precise connectivity Protein synthesis / folding GARS, BSCL2 DNA/RNA processing SETX, IGHMBP2 Axonal transport MFN2, DCTN1, DYNC1H1, BICD2 Neurite outgrowth Axonal guidance Cytoprotecion Cytoskeletal stability GARS, PLEKHG5, FBXO38 Proposed functions for genes in axonal CMT and distal HMN HSPB1, HSPB8, HSPB3 Cation-channel dysfunction ATP7A, TRPV4 L. Van Den Bosch and V. Timmerman (updated) Current Neurology and Neuroscience Reports (2006) Pareyson, Saveri and Piscosquito, Curr Mol Med (2014)
- 9. Mutations in small heat shock proteins associated with peripheral neuropathies Irobi et al. Nature Genetics (2004); Evgrafov et al. Nature Genetics (2004) Kolb et al., Neurology (2010); Nakhro et al., Neuromuscul Dis (2013) Rossor et al. JPNS (2012); Houlden et al. Neurology (2008) Holmgren, Bouhy, Timmerman (2012), eLS, John Wiley & Sons G 34RP39L E41K G 84R L99M R127WS135F R 136W /L R140G K141Q T151IS158fs E175XT180I R 188W P182L/S 88 168 P 1 2055 1915 PP 78 82 178 193 66 1431 150 R7S 93 170 196 K141E/N 24 87 IXI/VWDPF 117 151107 160 N-terminal domain α-Crystallin domain C-terminal end ACD ACD ACDβ4 β8 HSPB1 HSPB3 HSPB8 1 P P K141T Q 175X
- 10. Small HSPs are more than chaperones Almeida-Souza, Timmerman, Janssens (2011) BioArchitecture Holmgren, Bouhy, Timmerman (2012) eLS, John Wiley & Sons Protein folding Apoptosis Neuronal survival Cancer Alzheimer CMT small HSPs Translational controlCytoskeleton dynamics ALS Splicing regulation
- 11. HSPB1, HSPB5 and HSPB4 in human cancers: potent oncogenic role of some of their client proteins Arrigo & Gibert, Cancers (2014) Membrane signalling proteins Cytoskeleton, cell adhesion, tissue integrity Epithelial to mesenchimal transmission Gene expression Control of protein degradation Cell death, apoptosis, redox rate, aging Kinases, phosphatases, signal transduction Growth factors, receptors
- 12. HSPB8 (Hsp22) is involved in many different processes … regulating the proteolytic degradation of unfolded proteins (Huntington, AD, desmin-related cardiomyopathy) Holmgren, Bouhy, Timmerman (2012) eLS, John Wiley & Sons Role in CMT ? G93A-SOD1 expression induces a robust autophagic response specifically in muscle Crippa et al. (2013) Frontrier in Cellular Neuroscience
- 13. M. Coleman, Nature Review Neuroscience (2005) Mutant HSPB8 (K141E) Irobi et al. Human Molecular Genetics (2010) Mutant HSPB8 causes motor neuron-specific neurite degeneration
- 14. Mutant HSPB8 causes motor neuron specific neurite degeneration in axonal CMT 0 10 20 30 40 50 60 70 80 90 100 0 500 1000 1500 2000 2500 percentageofcells neuritelenght (µm) Control GFP HSPB8 K141N K141E Irobi et al. Human Molecular Genetics (2010)
- 15. Mutant HSPB8 causes protein aggregates in dermal fibroblasts Irobi et al. Neuromuscular Disorders (2012) HSPB8-K141N scale bar = 20 µm
- 16. Mutant HSPB8 protein aggregates recrute the cellular machinery to eliminate refolded or degraded misfolded HSPB8 proteins Scale bar = 10 µm Patient and control fibroblast cells were immunostained with antibodies against HSP70 (green) or Ubiquitin (green) and HSPB8 (red). HSPB8-K141N
- 17. Mutant HSPB8 causes reduced mitochondrial membrane potential in dermal fibroblasts Loss of mitochondrial membrane potential in patient’s fibroblasts. ∆Ψm was measured in cultured patients and controls fibroblasts. A: Valinomycin treated cells were used as a positive control for loss of ∆Ψm. B and C: fluorescent lipophilic cationic dye (TMRM) histogram analysis of ∆Ψm. D: Statistical analysis (∆Ψm geometric mean and SD) was performed on 3 different experiments. Irobi et al. Neuromuscular Disorders (2012)
- 18. HSPB8 patient fibroblast cells do not elicit marked apoptosis Quantification of DNA fragmentation in patient and control fibroblasts Cell death assay of distal HMN patients and controls fibroblast cells Irobi et al. Neuromuscular Disorders (2012) Caspase-Glo 3/7 assay (Promega) DNA fragmentation assay (BioVision)
- 19. Carra et al. JBC (2010) gmr-GAL4-UAS-SCA3(78Q)/+ gmr-GAL4-UAS-SCA3(78Q)/UAS-HSPB8#2 Wt K141E K141N K141N Quantification of eye degeneration in flies overexpressing either SCA3(78Q) alone or in combination with wild-type or mutated, K141E or K141N, HSPB8 Total number of eyes scored: 200-300 * = p< 0,05 as compared to SCA3; average values ± s.e.m. of n = 3 independent experiments. The K141E mutation significantly affects HSPB8 protective effect on SCA3- induced eye degeneration Drosophila ortholog of HSPB8: Implication of HSPB8 loss-of-function in protein folding disease The small HSP chaperones can impede protein aggregation in poly-Q disease
- 20. Hspb8-K141N knock-in and Hspb8 knock-out
- 21. HSPB1 (Hsp27) is involved in many different processes … interferes with the cell death signalling pathways at various levels Holmgren, Bouhy, Timmerman (2012) eLS, John Wiley & Sons Role in CMT ?
- 22. Accumulation of cytoplasmic aggregates in N2a cells transfected with mutant HSPB1 (HSP27) a-crystallin
- 23. HSPB1-mediated themotolerance 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 30min 60min 90min 0.2 0.4 0.6 0.8 1.0 WT R127W S135F R136W T151I S156Y P182L EGFP WT R127W S135F R136W T151I S156Y P182L EGFP WT R127W S135F R136W T151I S156Y P182L EGFP 30min 60min 90min Cell line Time at 45°C * ** ** * * * * * Relativesurvival Almeida-Souza et al. JBC (2010) SH-SY5Y cells stably expressing HSPB1 wt or mutants No Heat Shock 90 / 60 / 30 min Time at 45°C 48h recovery Cell counting
- 24. HSPB1 mutants present higher chaperone activity 0 0 .1 0 .2 0 .3 0 .4 0 .5 0 .6 W T R1 2 7 W S 1 3 5 F R1 3 6 W T 1 5 1 I S 1 5 6 Y P 1 8 2 L E G F P 0.1 0.2 0.3 0.4 0.5 0.6 ** * * ** * ** * ** * HS HR Relativechaperoneactivity WT R127W S135F T151I S156Y P182L EGFPR136W Almeida-Souza et al. JBC (2010) SH-SY5Y cells stably expressing HSPB1 and luciferase No Heat Shock Heat Shock 30min/44°C Heat Shock 30min/44°C Recovery: 3hs Luciferase activity Cycloheximide NO Recovery
- 25. Search for interacting partners to small HSP genes Cells expressing wild-type gene Cells expressing mutated gene prCMV TAPtag pA prCMV TAPtag pAWT CMT gene Mutant CMT gene Complex purification (TAP) “Wild-type complex” “Mutant complex” prCMV TAPtag pA prCMV TAPtag pAWT CMT gene Mutant CMT gene WT MUT m/z MS Differential interacting partner identification
- 26. Overactive HSPB1 mutants present a higher affinity to their client proteins Almeida-Souza et al. JBC (2010) Size Exclusion ChromatographyDifferential HSPB1 protein interactions
- 27. Increased monomerization of mutant HSPB1 leads to protein hyperactivity
- 28. Hyperactive HSPB1 mutants bind to tubulin and microtubules Almeida-Souza et al. Journal of Neuroscience (2011) IP from HEK293 cells IP from HEK293 protein extracts Mouse model
- 29. Co-localization of mutant HSPB1 to microtubules Almeida-Souza et al. Journal of Neuroscience (2011) Cos1 cells Normalized fluorescence intensity profiles Mutant HSPB1 disturb MT dynamics EB1-GFP tracking confirms slower MT polymerization Mutant HSPB1 stabilize MTs in vitro
- 30. Looking at the HSPB1 effect on individual MTs TUBB3-GFP EB1-GFP Microtubule plus ends Overactive HSPB1 disturb MT dynamics EB1-GFP tracking confirms slower MT polymerization Overactive HSPB1 stabilize MTs in vitro Hela Almeida-Souza et al. Journal of Neuroscience (2011)
- 31. HSPB1 facilitates the formation of non-centrosomal microtubules Almeida-Souza et al. PlosOne (2013) HSPB1 facilitates the formation of MTs in vitro and binds to MTs at early stages of their polymerization
- 32. Non-centrosomal -tubulinγ x x α-tubulin β-tubulin WT HSPB1 Mutant HSPB1 HSPB1 facilitates the formation of non-centrosomal microtubules Almeida-Souza et al. PlosOne (2013)
- 33. HSPB1 transgenic models for distal HMN/CMT2F d’Ydewalle et al. Nature Medicine (2011)
- 34. Almeida-Souza et al. Journal of Neuroscience (2011) MT stability is confirmed in HSPB1 transgenic mice HSPB1 mouse Disturbed axonal transport defects in symptomatic HSPB1 mutant mouse neurons
- 35. Decreased acetylated tubulin levels in symptomatic HSPB1 mutant mouse d’Ydewalle et al. Nature Medicine (2011) ***p<0.0001
- 36. HDAC6 is a microtubule-associated deacetylase HDAC6 : internal duplication of two class II catalytic domains + Ub binding domain Hubbert et al. Nature (2002) •Acetylated tubulin is most abundant in stable microtubules but is absent from dynamic cellular structures such as neuronal growth cones. •Decrease in HDAC6 increases tubulin acetylation stable microtubules •Inhibition of HDAC6 improves the dynamics of tubulin improve axonal transport
- 37. Treatment significantly increased motor performance of HSPB1_S135F mice HDAC6 inhibitors rescue axonal transport defects and restore the distal HMN/CMT2F phenotype Treatment increased electrophysiological parameters such as CMAP and SNAP d’Ydewalle et al. Nature Medicine (2011) ***p<0.0001
- 38. HDAC6 inhibitors reverse the axonal loss in mutant HSPB1 mice d’Ydewalle et al. Nature Medicine (2011) Treatment leads to muscle re-innervation and rescues axonal transport *** p<0.0001
- 39. Microtubule dynamics in the PNS Almeida-Souza, Timmerman & Janssens, Bio-Architecture (2012)
- 40. Microtubule dynamics in disease
- 41. HspB1 deficient mice display impaired wound healing • HspB1 regulates inflammatory gene expression and drives cell proliferation. • The expression of HspB1 protein and mRNA is controlled by the cell cycle. • HspB1-deficient mice (genOway, LoxP flaking exon 1-3). • HspB1-deficient fibroblasts have: – increased expression of the pro-inflammatory cytokine, IL-6, and reduced proliferation – reduced entry into S phase and increased expression of Cdk inhibitors p27(kip1) and p21(waf1) • There was a significant impairment in the rate of healing of wounds in hspB1-deficient mice: – reduced re-epithelialisation and collagen deposition – increased inflammation • HspB1 deficiency augments neutrophil infiltration in wounds, driven by increased chemokine (C-X-C motif) ligand 1 expression. Crowe et al. PlosOne (2013)
- 43. P182L_HSPB1 disrupts neurofilament assembly and the axonal transport of specific cellular cargoes • P182L but not wt HSPB1 leads to formation of insoluble intracellular aggregates and to sequestration in the cytoplasm of selective cellular components, including NF-M and p150 dynactin Ackerley et al. HMG 2006;15:347-354 HSPB1wt HSPB1 (P182L) HSPB1-HA P182L-HA Anti-HA GFP
- 44. NFs are bounded to and transported by the motor proteins kinesin and dynein NFs detached from a motor protein, but can easily re-associate. NF phosphorylation state determines association with kinesin or dynein. NFs dissociated from the motors and restricted from re-association, represented as NF bundles Neurofilaments can exist in three distinct pools Holmgren, Bouhy, Timmerman JPNS (2012)
- 45. Proline-directed kinases and phosphatases in neurofilament phosphorylation Holmgren, Bouhy, Timmerman JPNS (2012)
- 46. HSPB1 mutations increase Cdk5 mediated phosphorylation‑ of neurofilaments Holmgren et al. Acta Neuropathologica (2013) Proximal Distal Wild type HSPB1 Mutant HSPB1 neurofilaments phosphorylated neurofilament kinesin dynein-dynactin complex microtubules Proximal Distal Silencing Cdk5 restores NF-kinesin interaction and NF solubility in HSPB1 mutant
- 47. Complex pathomechanisms of small HSP mutations stabilization of MT autophagy deficit sHSP properties axonal degeneration acetylation of MT sHSP functions axonal transport defect mitochondrial depletion protein aggregation redox iron metabolism ER stress chaperoning binding affinity oligomerization phosphorylation cytoskeleton apoptosis oxidative stress
- 48. HMSN hereditary motor and sensory neuropathy = ‘CMT’ Motor Sensory & Autonomic HSAN hereditary sensory & autonomic neuropathy HMN hereditary motor neuropathy Harding & Thomas, J. Med. Genet (1980), J. Neurol. Sci (1980) and Brain (1980) Harding, A.E. (1993) In: Dyck, Thomas, Griffin, Low and Poduslo (Eds) In: Peripheral Neuropathy Dyck, P.J. (2005) In: Peripheral Neuropathy Various kinds of inherited peripheral neuropathies
- 49. Pathomechanisms of HSAN genes Rotthier, Baets, Timmerman & Janssens, Nature Reviews Neurology (2012) + updates SCN9A/SCN11A Dystonin DST cytoskeleton linker protein ATL1 & ATL3
- 50. Overview of AD-HSAN subtypes Rotthier, Baets, Timmerman & Janssens, Nature Reviews Neurology (2012) Updated with: Leipold et al. AJHG (2013), Zhang et al. AJHG (2013), Kornak et al. Brain (2014) * Online Mendelian Inheritance in Man (OMIM) reference number ‡ Congenital onset in one patient with hypotonia, cataracts, microcephaly and vocal cord paralysis § Childhood onset in one patient HSAN SCN11A loss of pain perception (2 de novo), familial episodic pain (2 missense) Childhood 604385 HSAN ATL3 Loss of pain perception and destruction of pedal skeleton (1 missense in 2 families) Adolescence to adulthood 609369
- 51. HSAN I (CMT2B) caused by mutations in RAB7A Rotthier, Baets, Timmerman & Janssens, Nature Reviews Neurology (2012) Updated with: Cogli et al. Acta Neuropathologica (2013), … Decreased nucleotide affinity and deregulation of nucleotide exchange Subtle increase in duration of RAB7 association with target membranes Peripherin Altering neurofilament dynamics
- 52. Comprehensive map of the effectors of Rab GTPases Gillingham et al. Developmental Cell (2014)
- 53. Human L129F Rab7 mutation in Drosophila dWT Drosophila WT RAB7 hWT Human WT RAB7 hL129F Human mutant RAB7 0N3R tau TAU mutant painless Painless mutant TrpA1[1] TRPA1 mutant D42-Gal4 Motor neuron driver P0163-Gal4 Sensory neuron driver 221-Gal4 UAS-GFP Rab7 is a highly conserved protein; the human form shares 76% identity and 95% similarity with its single Drosophila orthologue We used double transgenic lines expressing Rab7 alleles from both chromosome 2 and 3
- 54. Mutant Rab7 larvae show decreased motor performance Distance travelled in two minutes by larvae Janssens et al. Neurobiology of Disease (2014)
- 55. Sensory performance is affected in mutant Rab7 larvae Thermotaxis assay Nociception assay „warm side (30°C)‟ „cold side (22°C = preferred )‟ soldering iron with a chisel-shaped tip at 43°C Janssens et al. Neurobiology of Disease (2014)
- 56. Transport of mutant Rab7+ vesicles is altered in sensory axons of 3rd instar larvae and in neurites of SH-SY5Y cells No changes in neuronal morphology Rab7+ vesicles in long axons of sensory ddaE and ddaD neurons of dissected larvae: No difference between the two genotypes with regard to speed and directionality of vesicle movement However; L129F Rab7 vesicles pause less than their wt counterparts ventral multi-dendritic sensory vpda neuron of hemisegment 6 Janssens et al. Neurobiology of Disease (2014)
- 57. CMT2B mutations in rab7 cause dosage-dependent neurodegeneration due to partial loss of function Cherry et al. eLIFE (2013)
- 58. Defective axonal transport of Rab7 results in dysregulated trophic signaling Zhang et al. J Neurosci. (2013) Rab7 mutants dysregulate axonal transport and diminish retrograde signaling of nerve growth factor (NGF) and its TrkA receptor. Rab7 mutants induce premature degradation of retrograde NGF-TrkA trophic signaling.
- 59. Stuck in traffic: an emerging theme in diseases of the nervous system Neefjes & van der Kant, Trends in Neuroscience (2014)
- 60. Conclusion: Pathogenic mechanisms for CMT genes Timmerman, Strickland, Züchner GENES (2014)