Download as PDF, PPTX


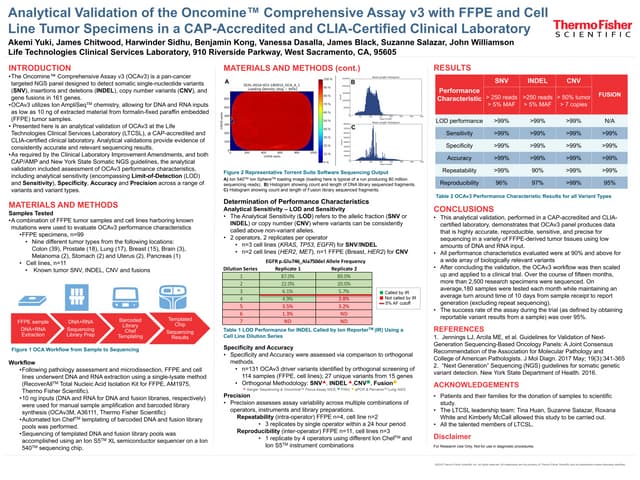
The document describes an Oncomine tumor mutation load assay that assesses tumor mutational load in FFPE samples using a targeted Ion Ampliseq panel covering approximately 1.7 Mb of genomic DNA. The assay is optimized for speed and reproducibility, allowing for detailed reports on mutation counts and signatures that correlate well with whole exome sequencing. This research tool may aid in identifying predictive biomarkers for cancer immunotherapy and improve the understanding of responses to immune checkpoint blockade inhibitors.