Downloaded 76 times


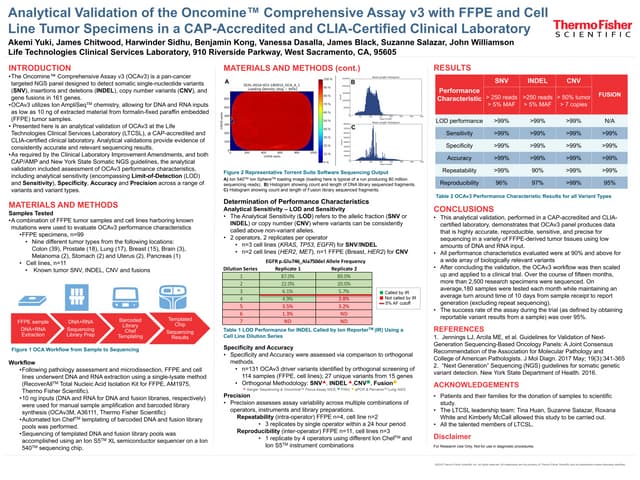
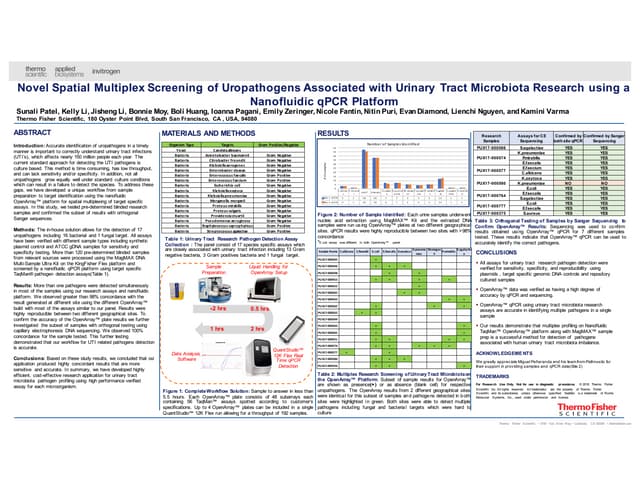
This document details the development and validation of Oncomine cfDNA assays for noninvasive detection of low-abundance tumor mutations from blood samples, achieving a sensitivity of over 80% and specificity exceeding 98% at a 0.1% limit of detection. Three distinct panels were created for lung, colon, and breast cancers, each targeting specific mutation hotspots. These assays support comprehensive oncology research by enabling detection of rare mutations that may inform tumor progression and therapeutic resistance.