Downloaded 36 times




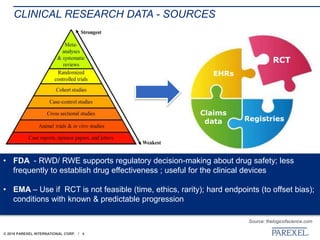
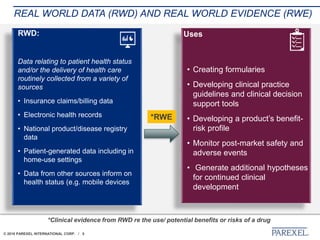
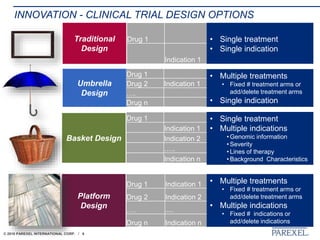

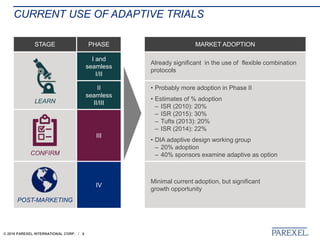
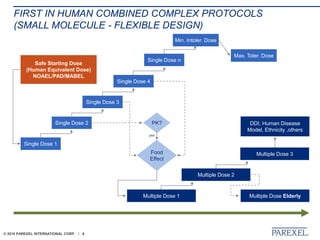
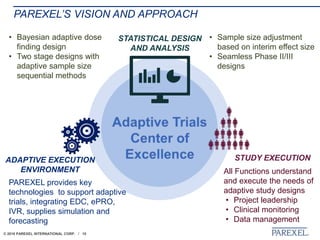


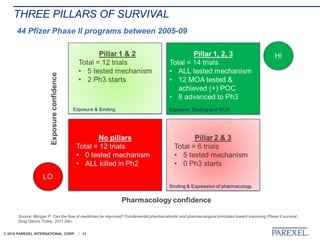

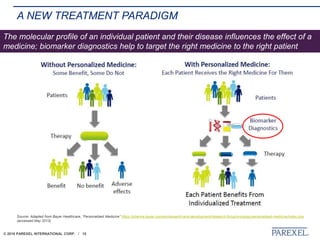

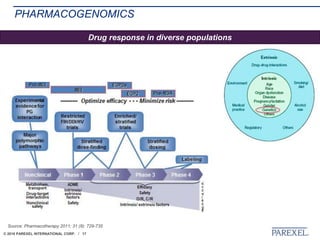
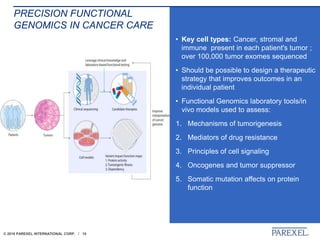


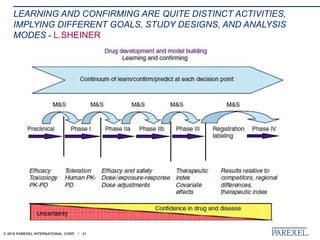
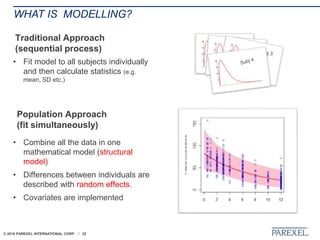


The document discusses trends in early drug development, focusing on innovative trial designs, the use of genomic and other biomarkers in clinical trials, and the modeling and simulation processes in these contexts. It emphasizes the importance of adaptive trials and the integration of real-world data in regulatory decision-making to improve drug safety and effectiveness. Additionally, it highlights how personalized medicine aims to tailor treatments based on individual genetic profiles to enhance clinical outcomes.

































![Dancey Clinical Trials Vancouver Dancey 20110302 Final.Ppt [Compatibility Mode]](https://cdn.slidesharecdn.com/ss_thumbnails/danceyclinicaltrialsvancouverdancey20110302finalpptcompatibilitymode-13018535926617-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)















































