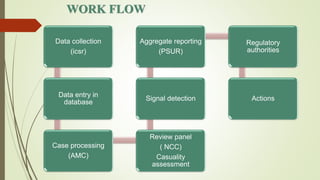
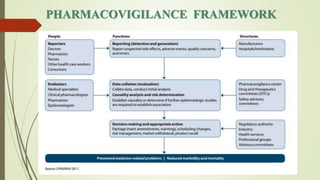
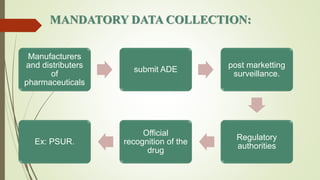
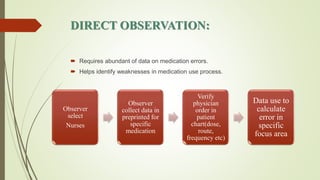
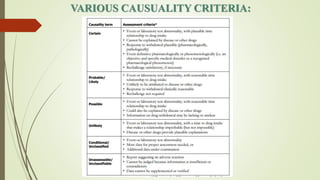
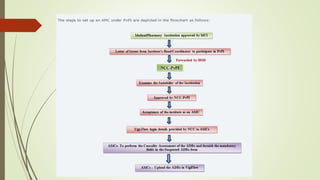
Pharmacovigilance is the science of monitoring approved drugs to detect adverse effects. It aims to identify new risks, assess known risks, and prevent harm. Pharmacovigilance relies on collecting data on adverse drug reactions (ADRs) through passive and active methods. Data is analyzed to detect safety signals and assess risks and benefits of medicines to optimize safe use. Regulatory authorities use pharmacovigilance data to take actions like updating product information or withdrawing approval if risks outweigh benefits.