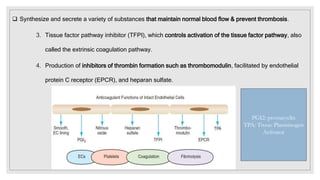
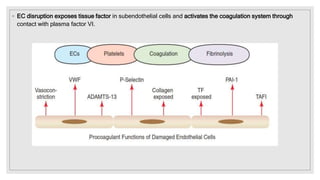
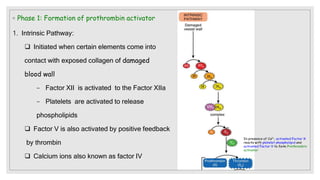
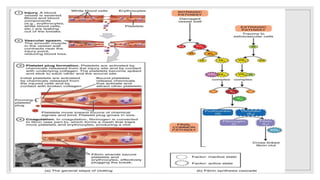
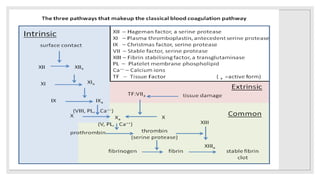
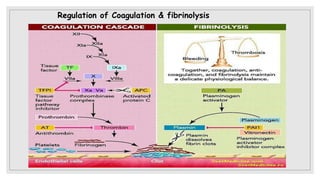
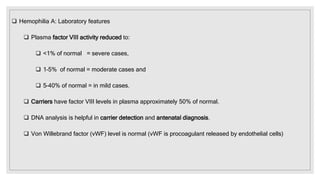
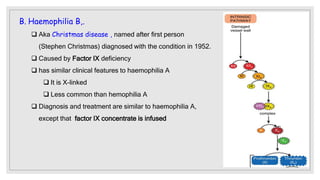
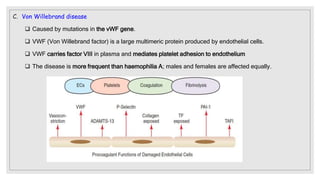
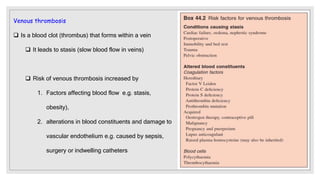
The document discusses the complex physiological process of haemostasis, which includes coagulation, fibrinolysis, and vascular reconstruction. It covers the mechanisms involved in maintaining blood fluidity, the causes and types of coagulation disorders, and the treatments for conditions such as hemophilia and von Willebrand disease. Additionally, it examines the balance between coagulation and anticoagulation within the vascular system and describes both inherited and acquired disorders of coagulation.