Download as PDF, PPTX





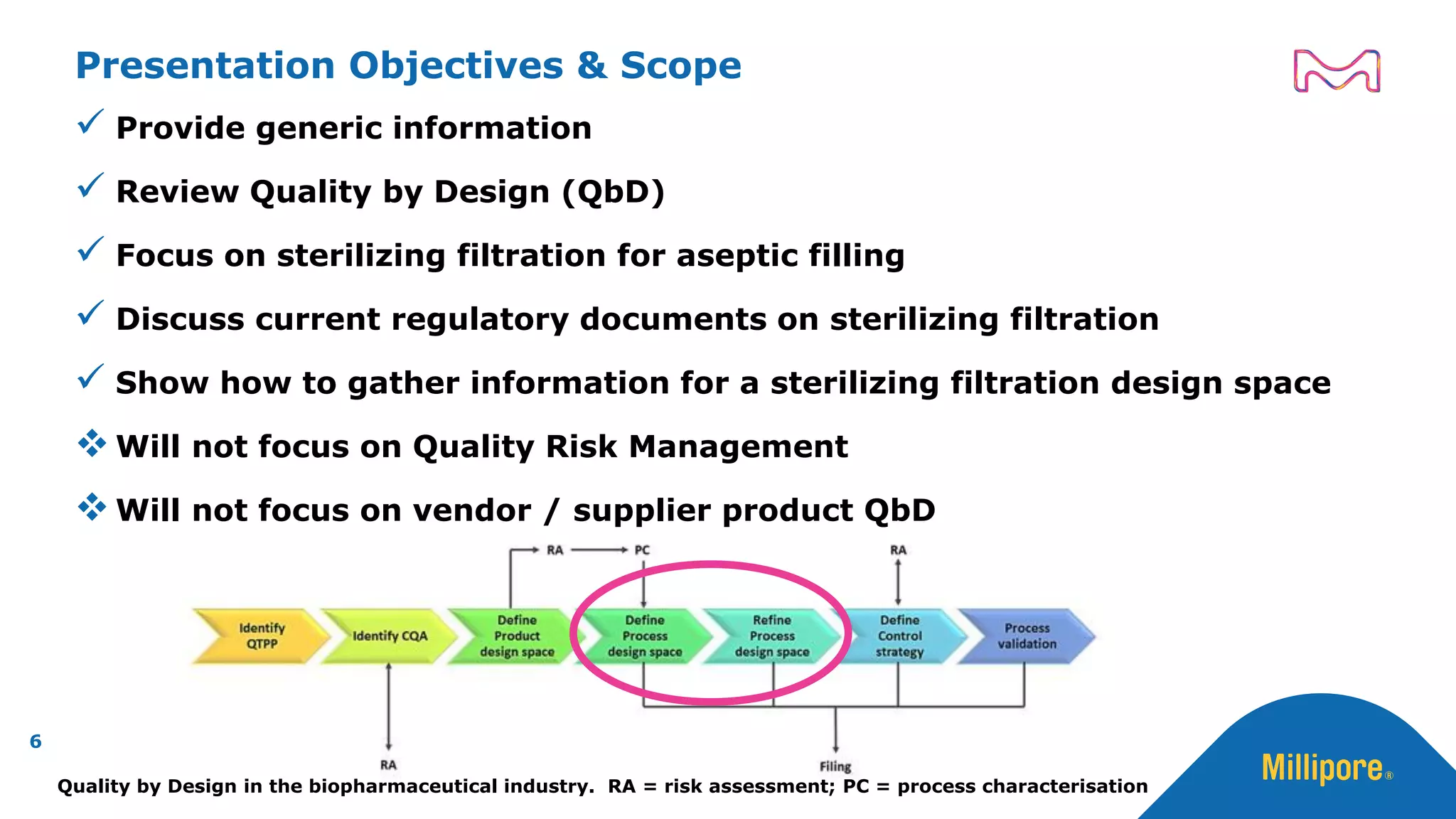


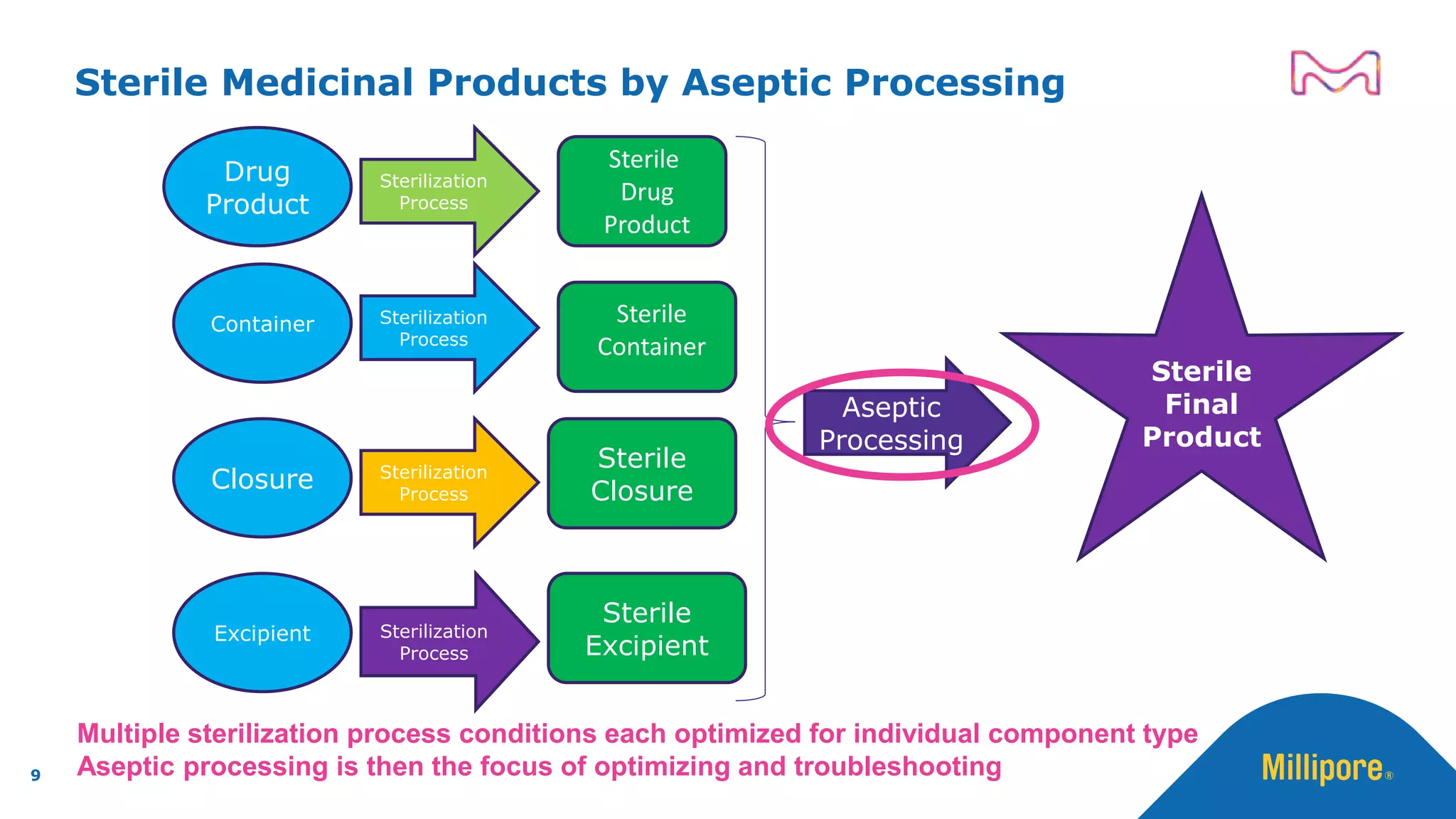
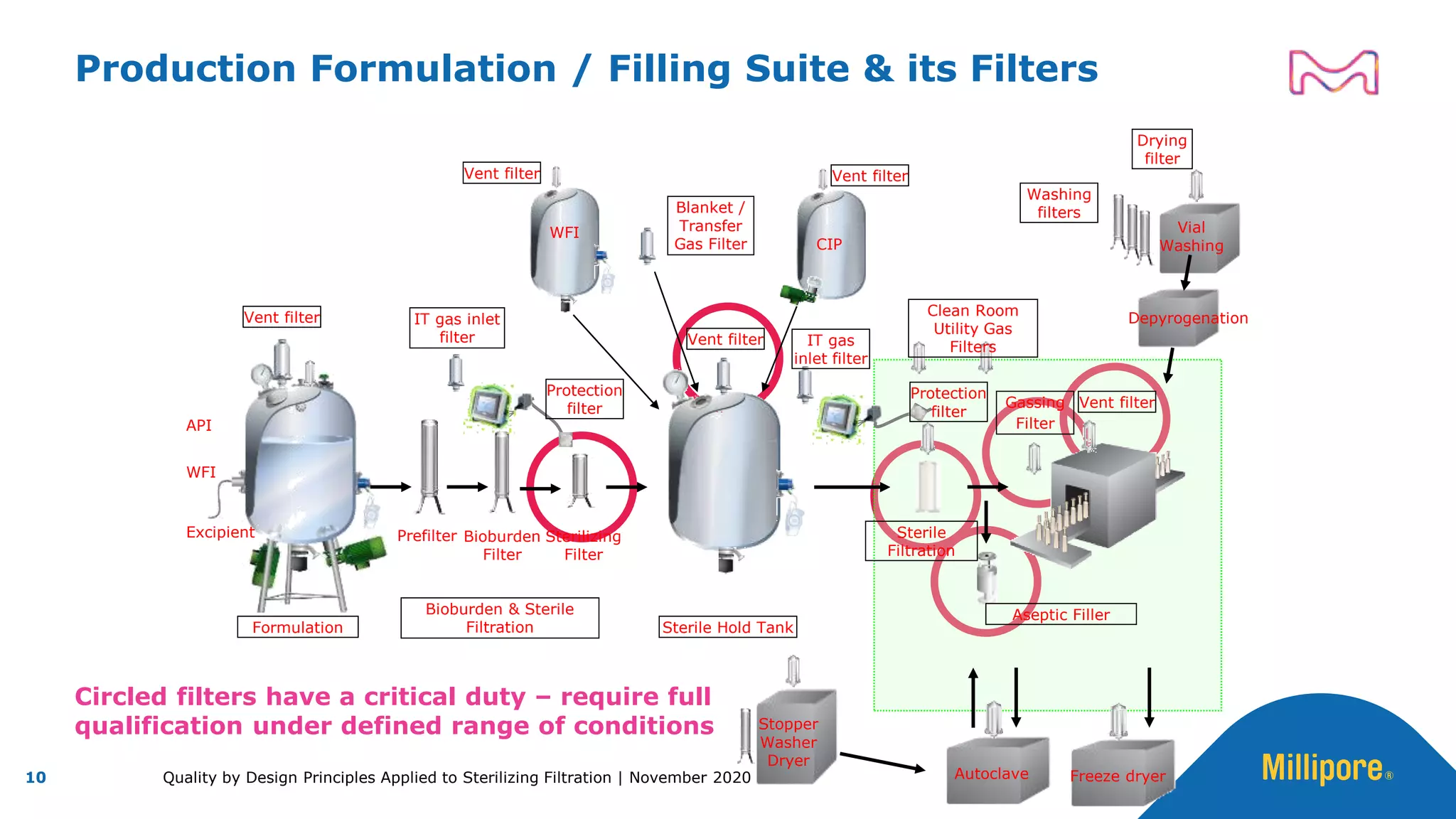

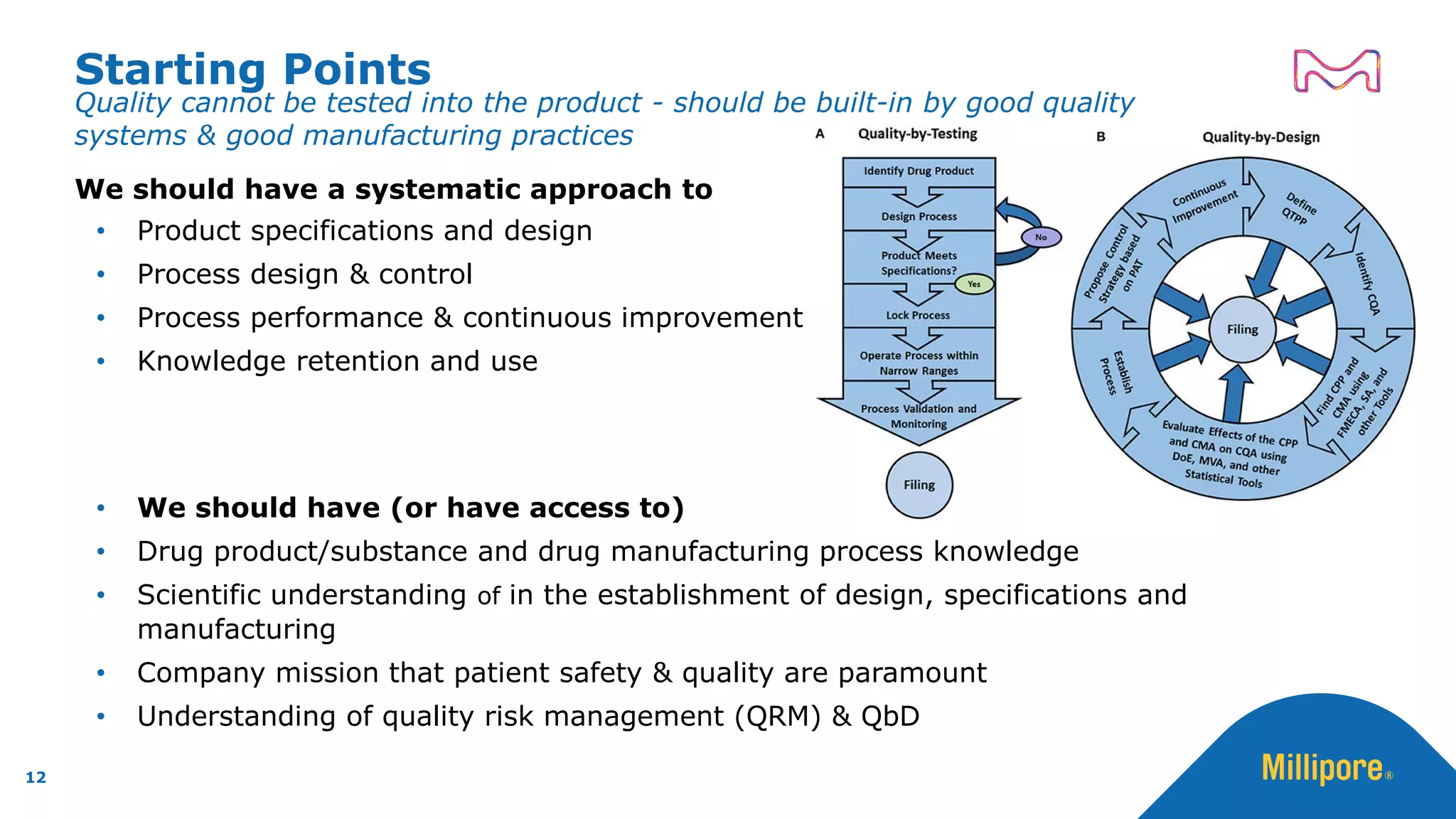

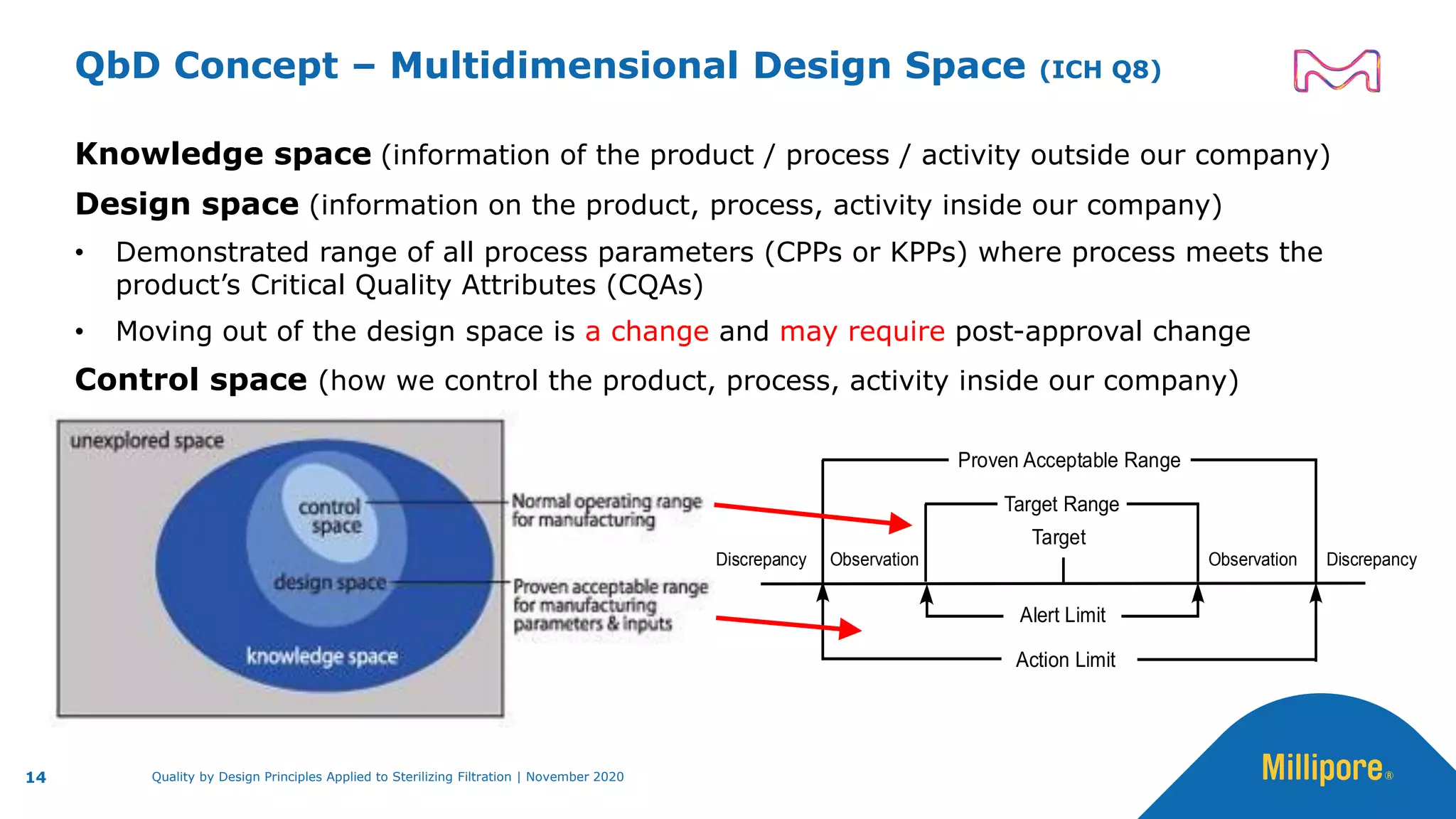
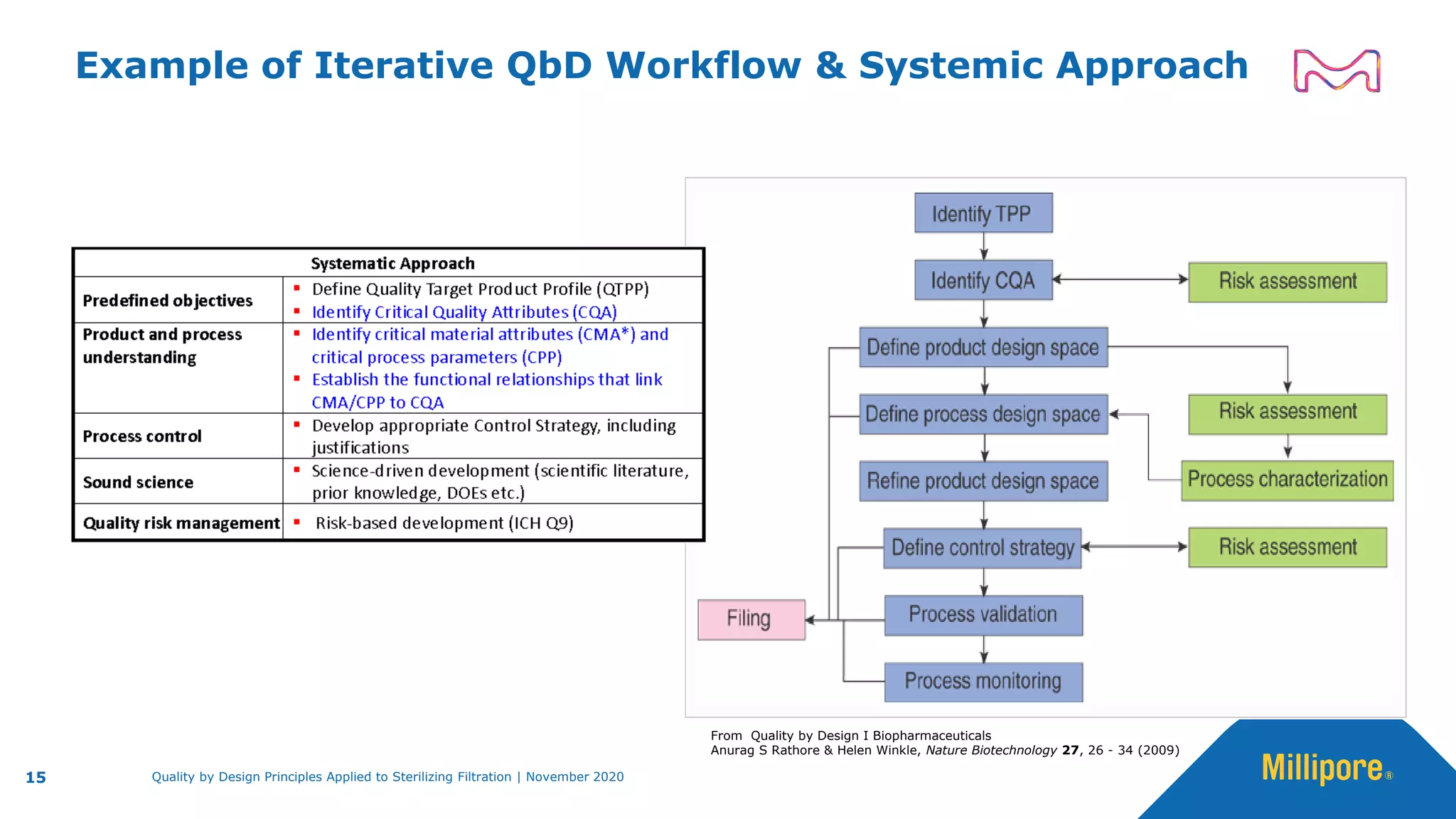

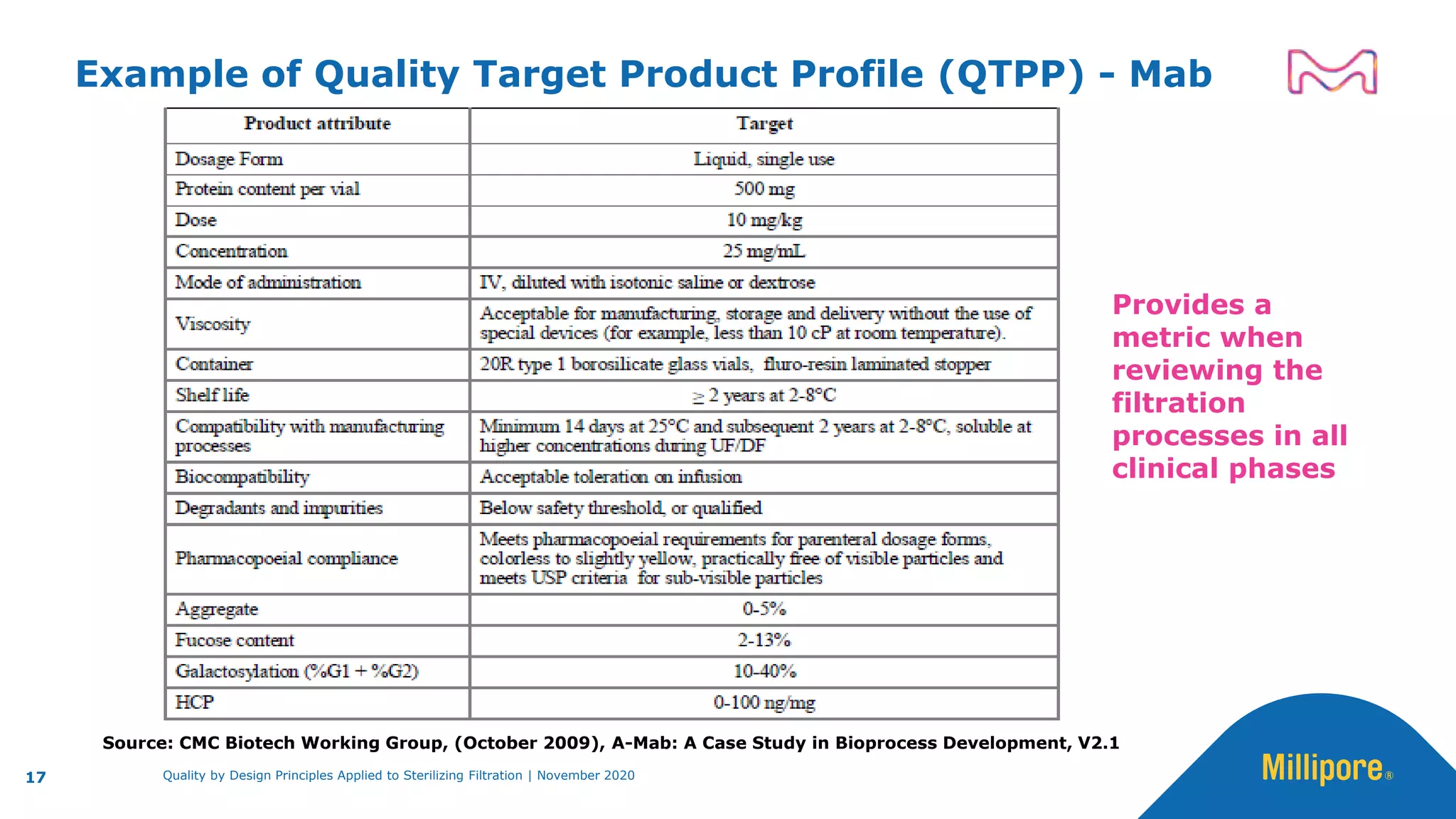
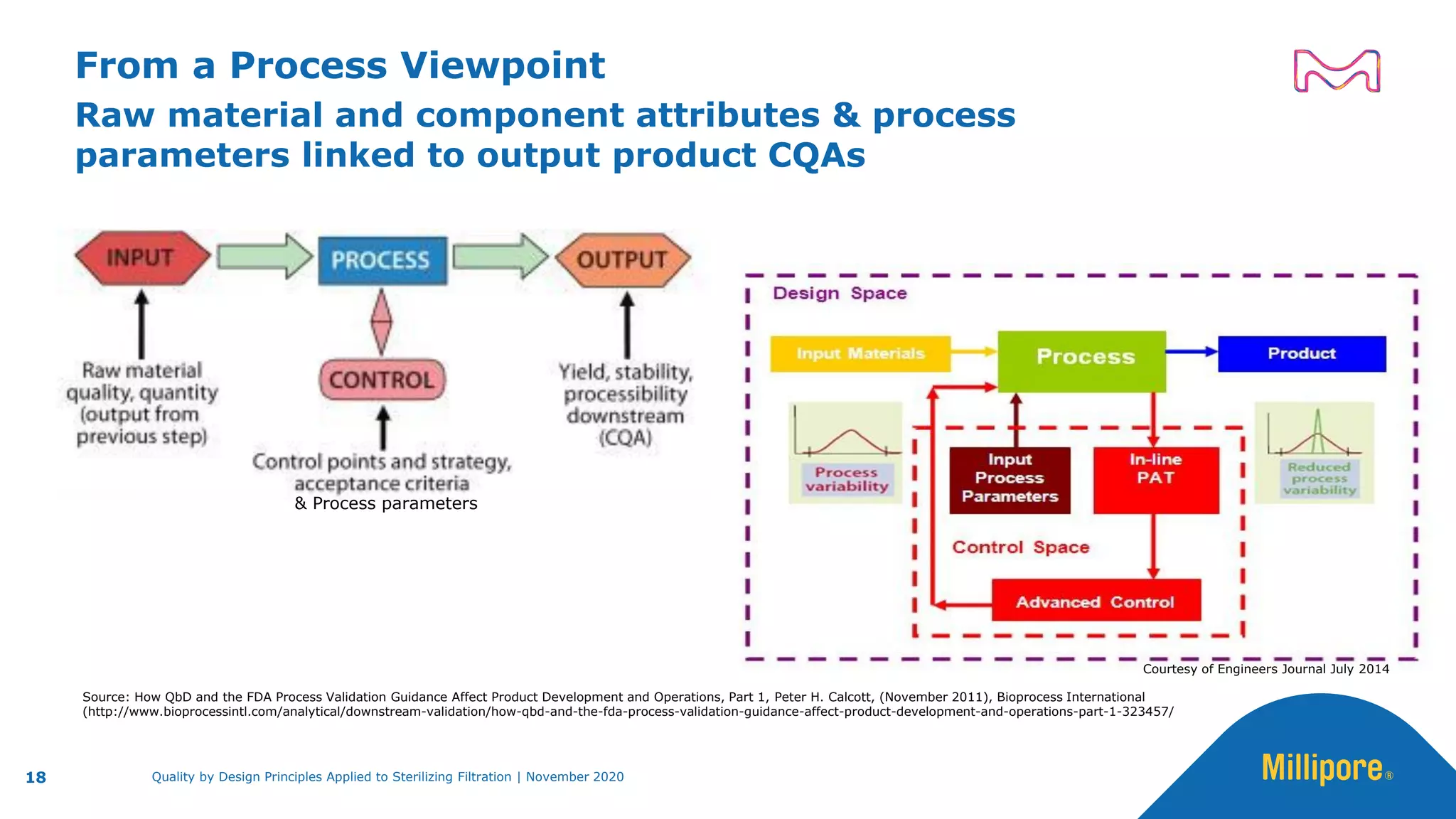






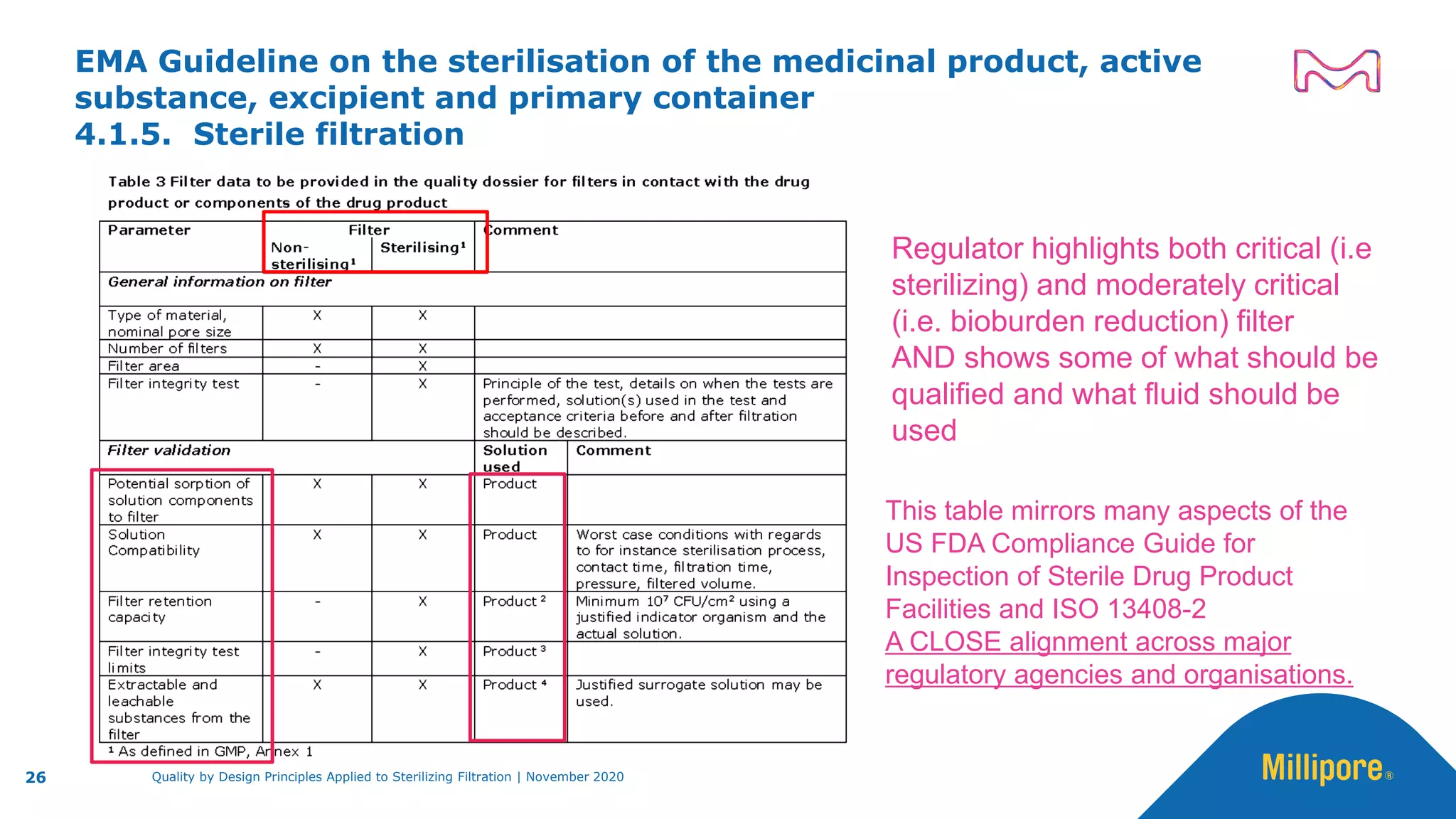






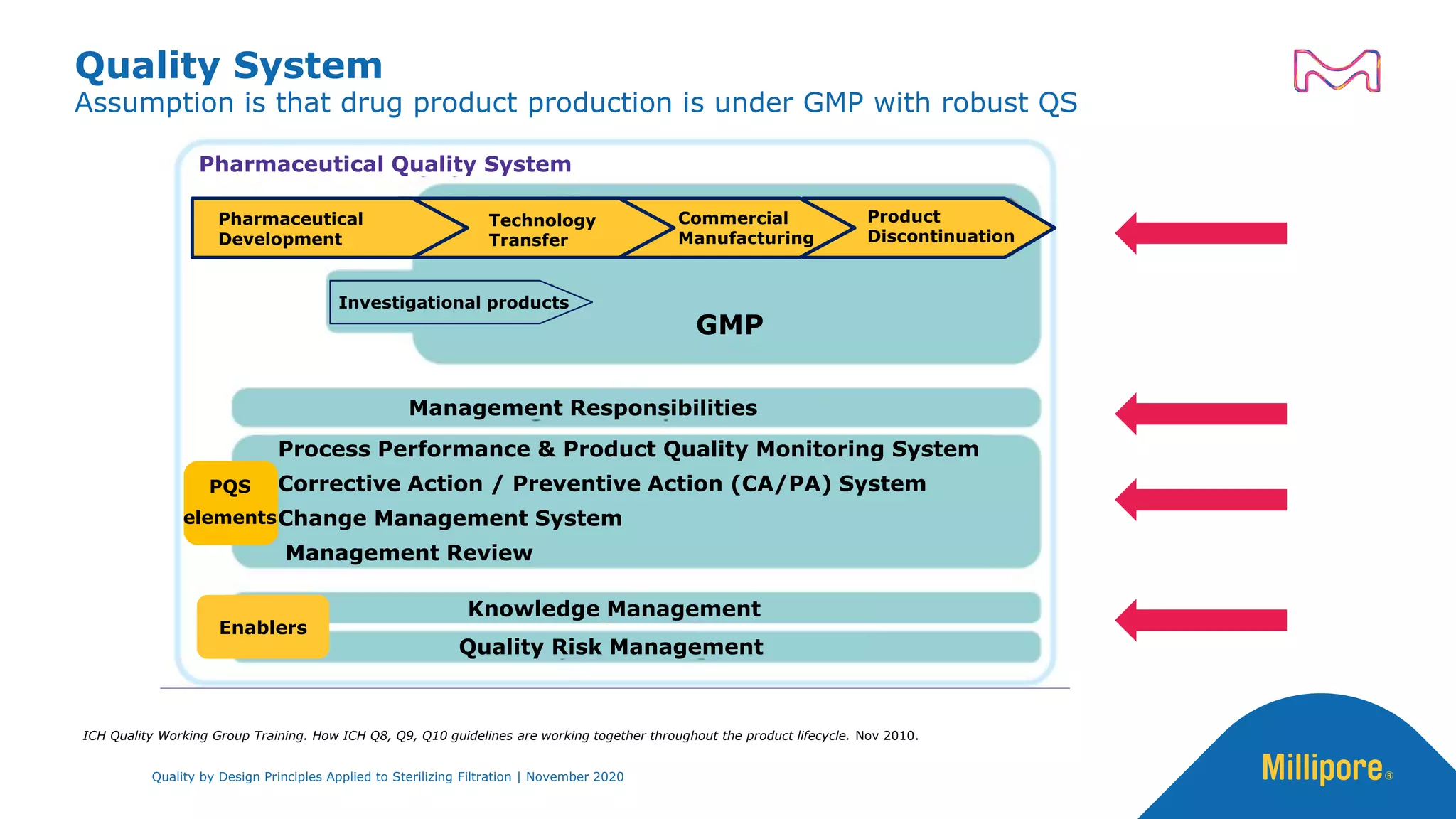

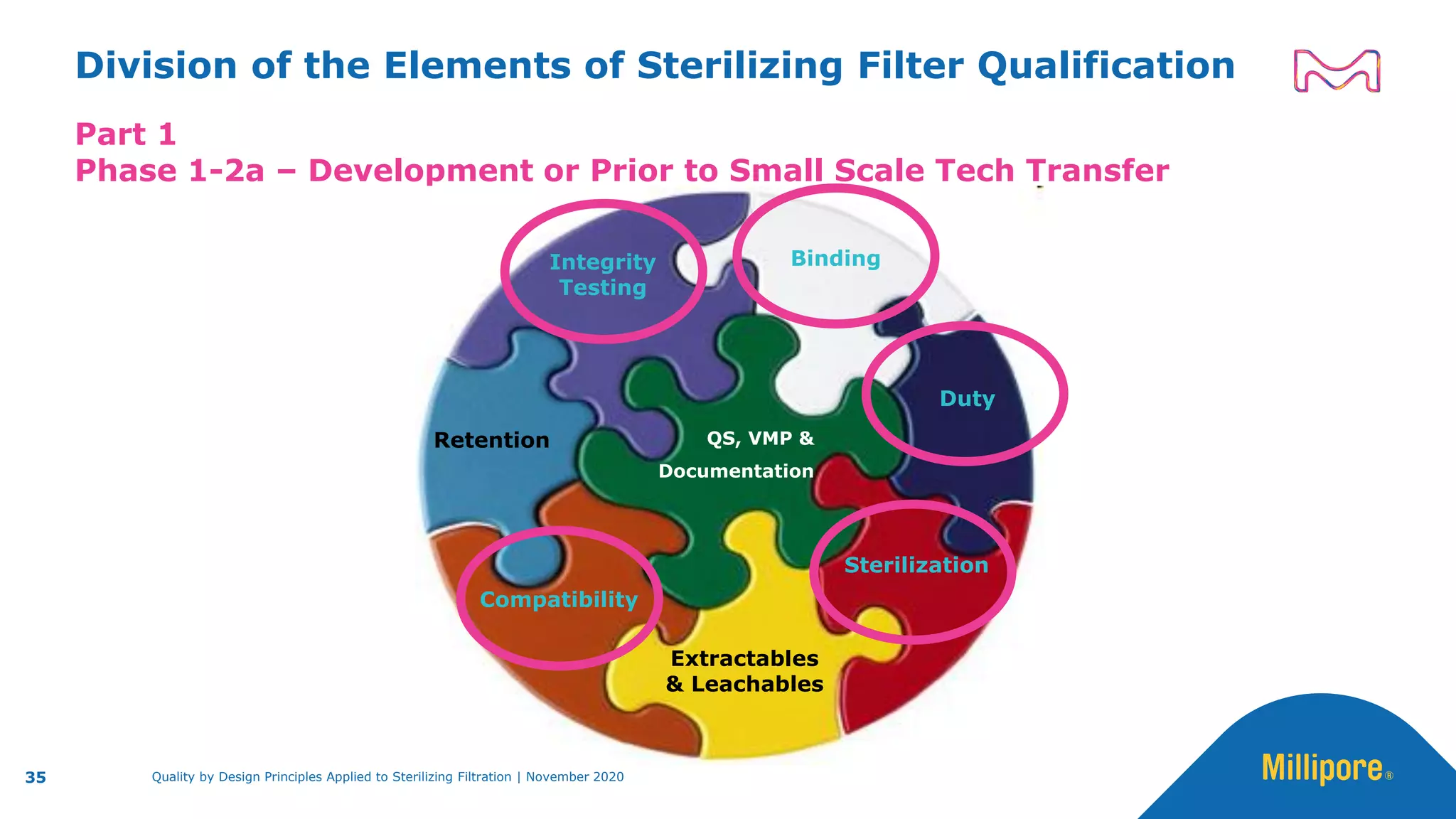








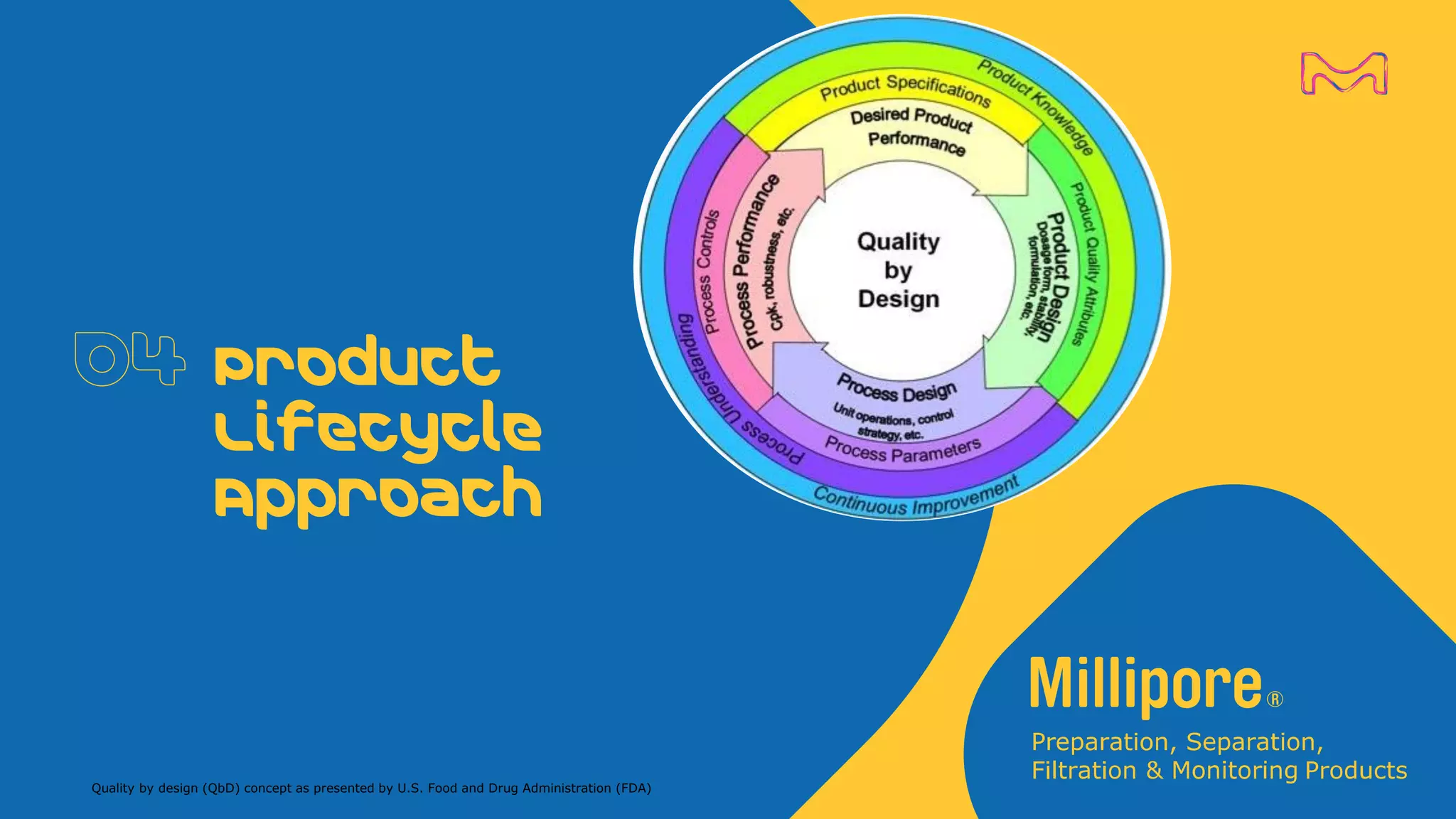


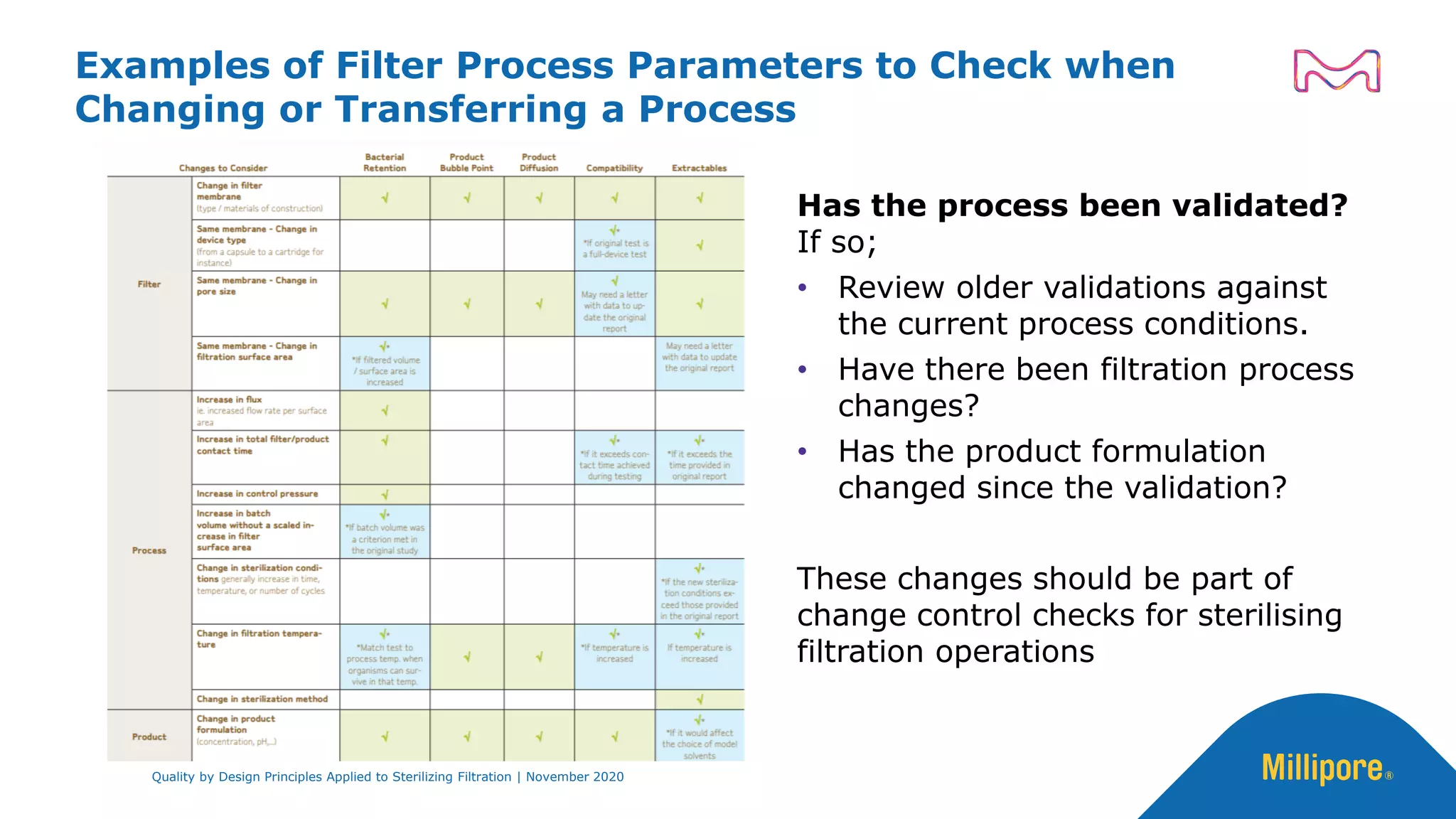







The document discusses the application of Quality by Design (QbD) principles to sterilizing filtration in the biopharmaceutical industry, emphasizing its importance for ensuring drug product quality and patient safety. It outlines concepts, regulatory guidance, and key aspects related to sterilizing filter qualification, as well as iterative workflows and design spaces necessary for efficient filtration processes. The presentation highlights the necessity of integrating QbD throughout the product lifecycle to achieve compliance with evolving regulatory requirements.























































































