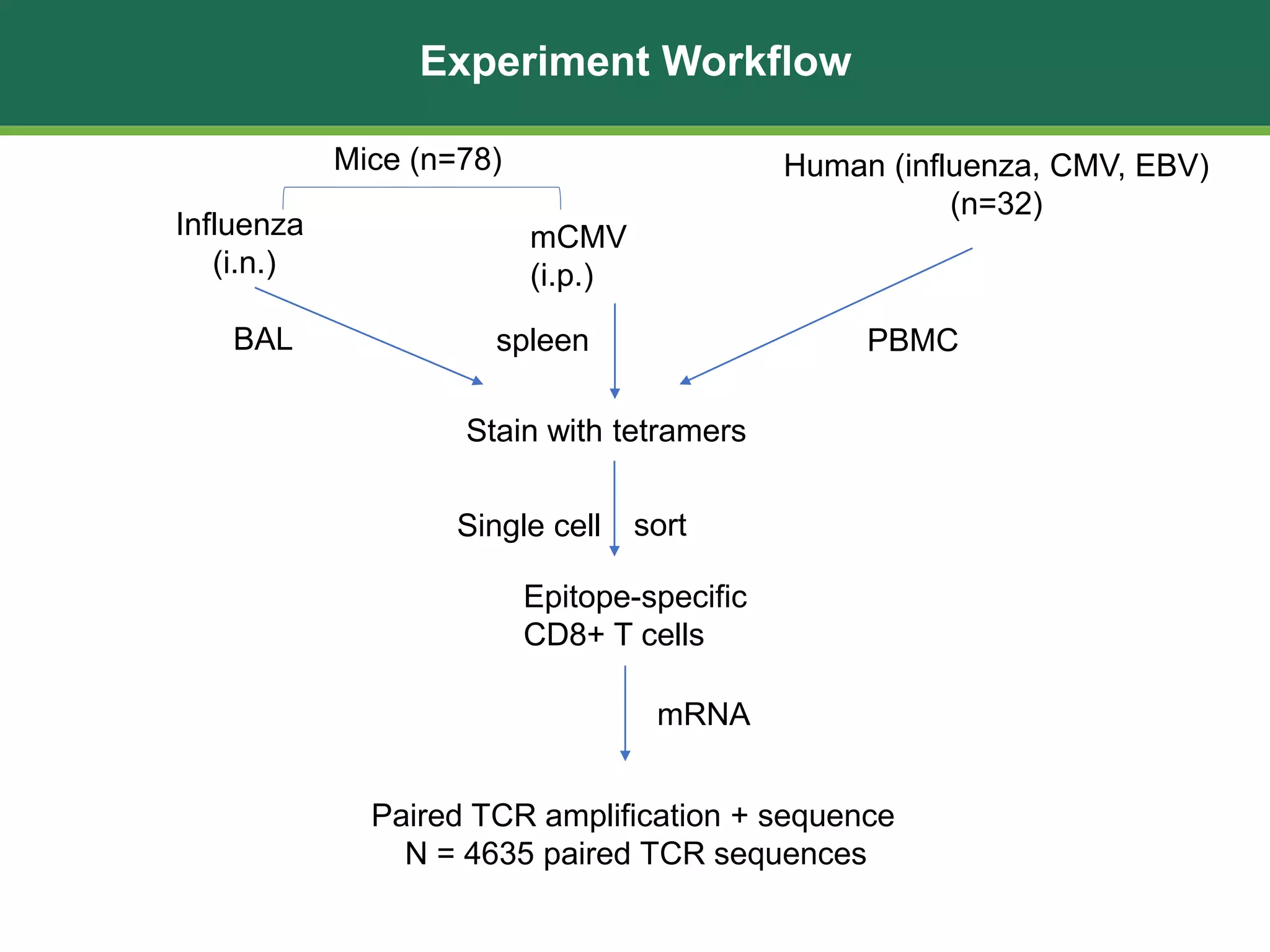
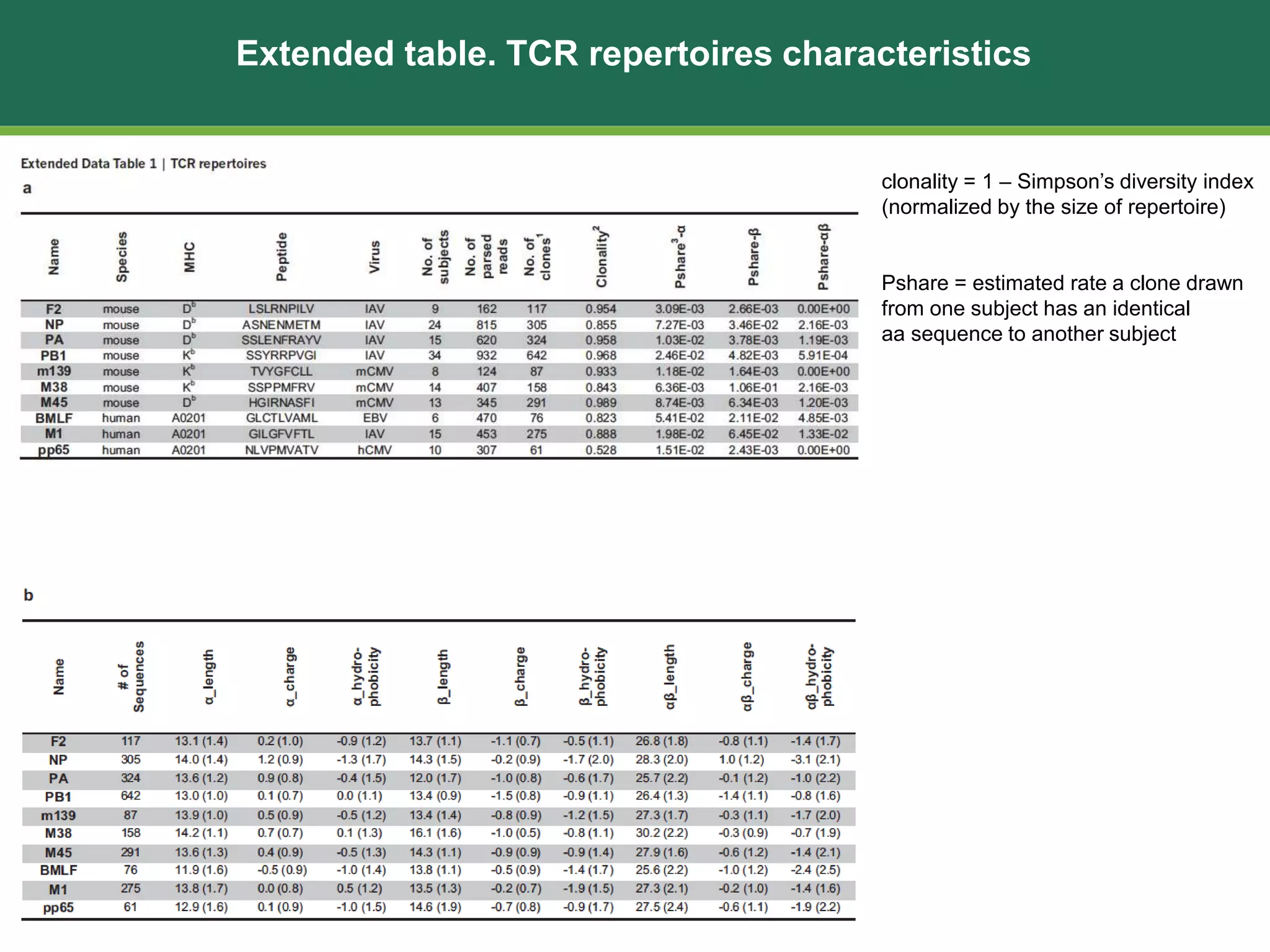
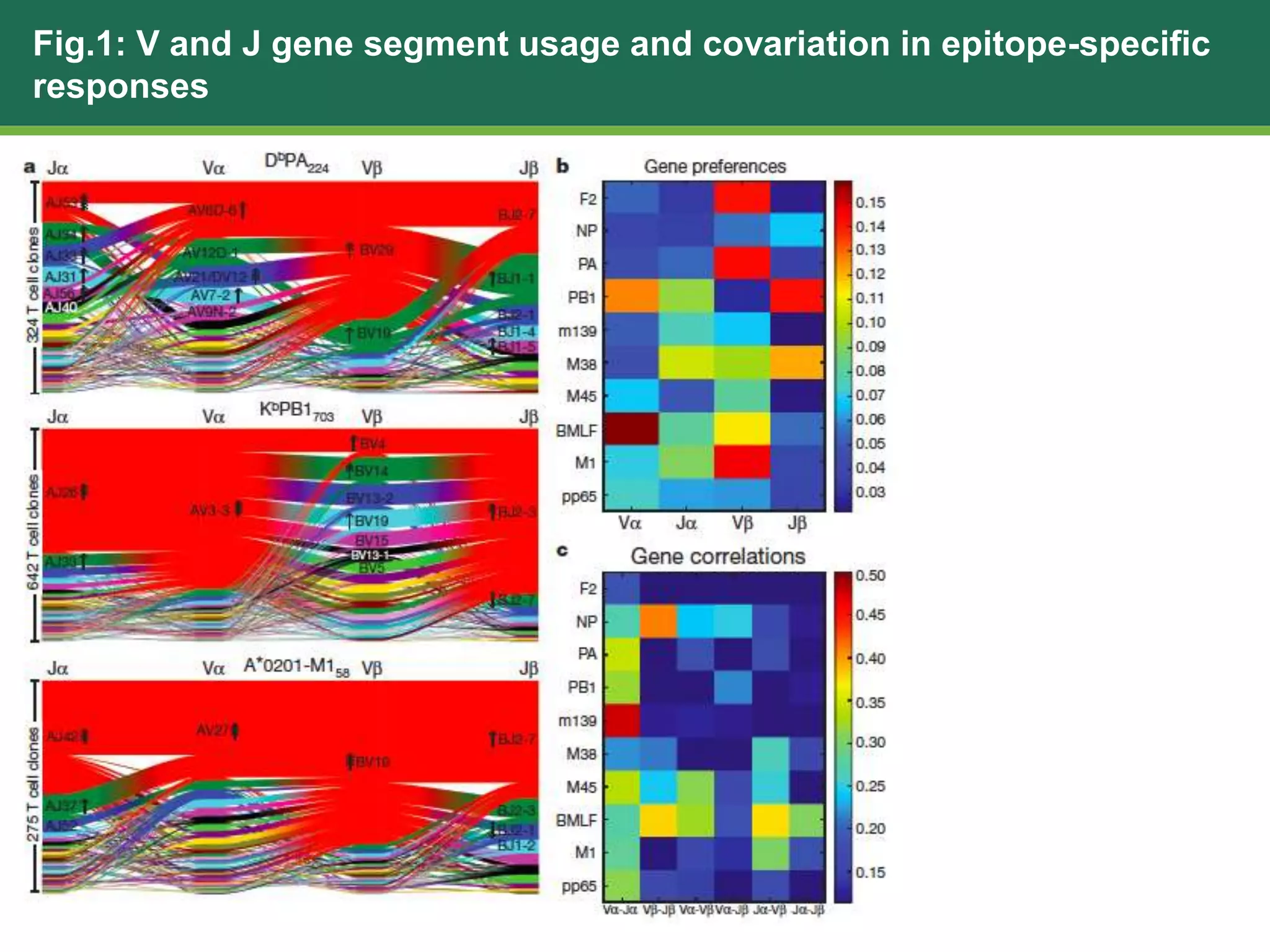
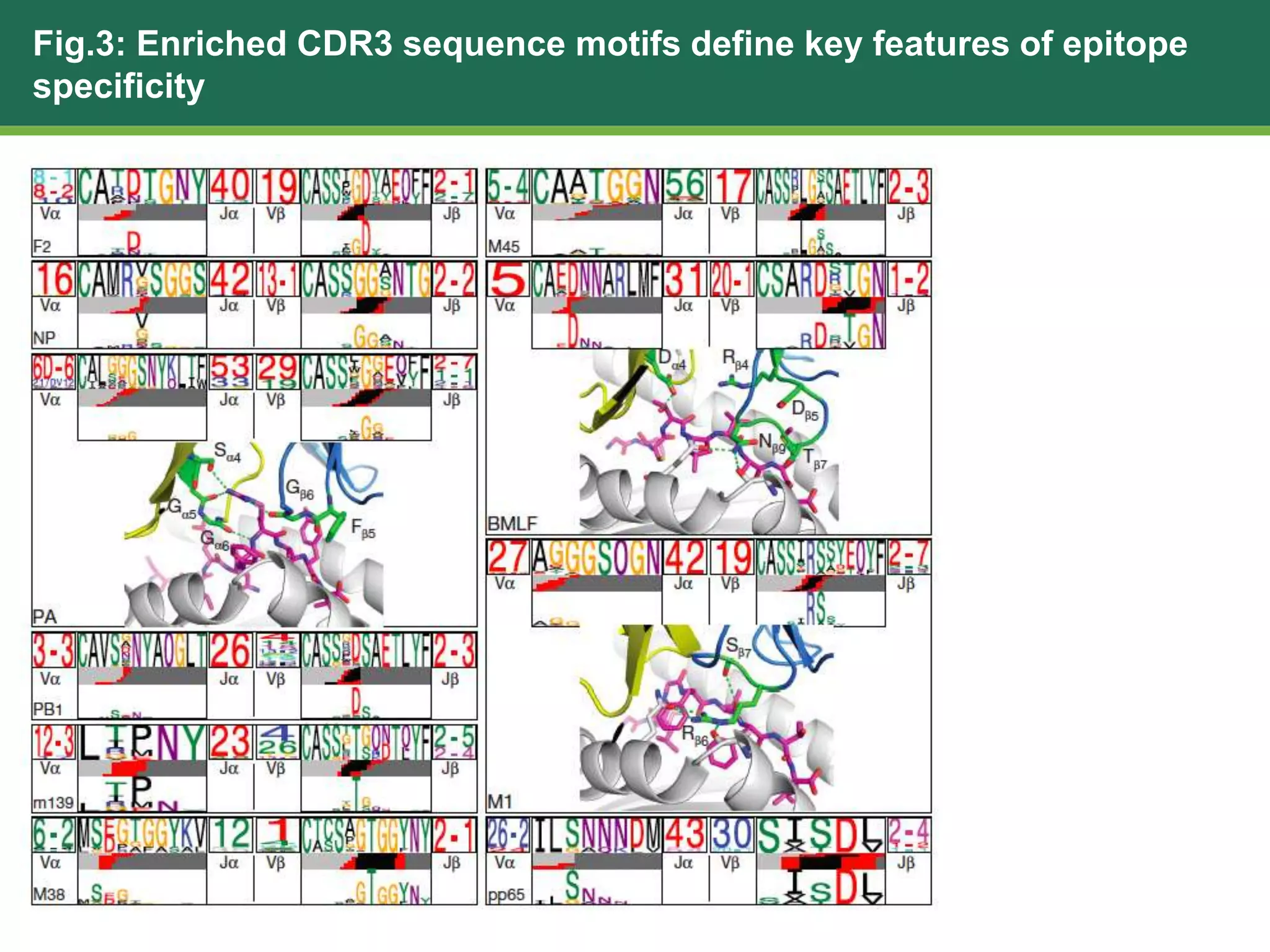
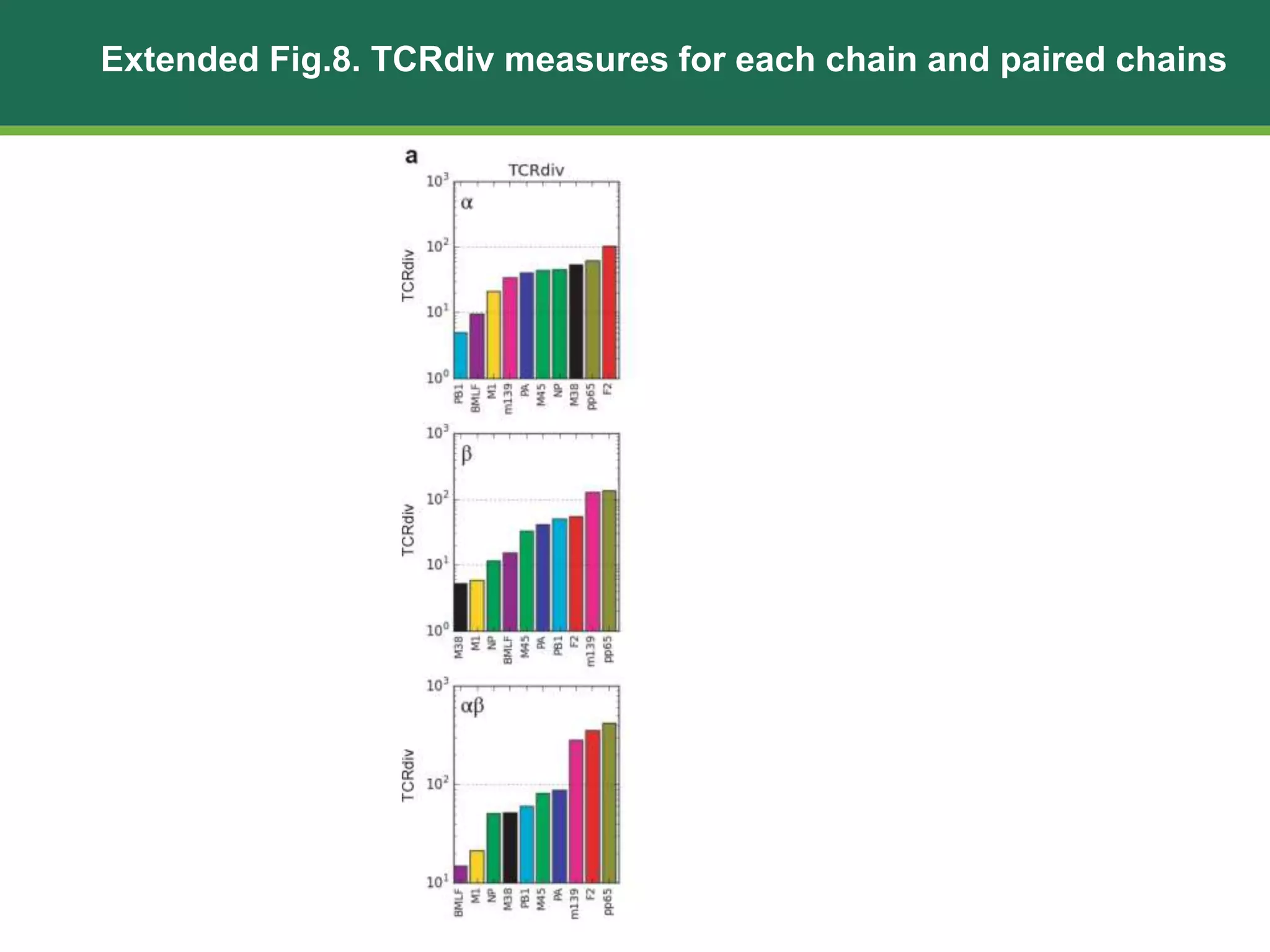
This document summarizes the characterization of 10 epitope-specific CD8+ T cell receptor repertoires from over 4,600 single cells. Key findings include quantifying gene segment usage, epitope selection, TCR similarity using TCRdist, repertoire diversity with TCRdiv, and developing a distance-based classifier to assign unobserved TCR. The work demonstrates that predictive models for TCR-pMHC recognition may be possible despite tremendous TCR diversity, with potential applications to analyze clinical T cell receptor repertoire data.