Hutchinson-Gilford Progeria Syndrome (HGPS) is a rare genetic disorder characterized by accelerated aging due to a mutation in the LMNA gene, leading to the production of the abnormal protein progerin. Symptoms develop in early childhood, resulting in severe growth retardation and complications that often lead to death by age 14. Current treatment options are limited and focus on supportive care and experimental therapies, including gene therapy and drugs targeting specific disease manifestations.

![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1226
1) INTRODUCTION
Progeria is also known as Hutchinson– Gilford Progeria Syndrome it is a rare, sporadic,
monogenic, autosomal dominant, fatal childhood disease that belongs to a group of ailment
called as laminopathies which affect nuclear lamins.[1,2,3]
Progeria was first portrayed in 1886
by Jonathan Hutchinson.[1]
It was additionally depicted autonomously in 1897 by Hastings
Gilford. The condition was later named Hutchinson-Gilford Progeria Syndrome (HGPS).[2]
The word progeria comes from the Greek words "pro" meaning "before", and "geras"
meaning "old age".[2]
In this disease the aging process of the body surge much more faster
than what it does in normal individuals. This process of aging gallops to about seven times
the conventional rate. Because of this accelerated aging, a child of ten years would have a
look of 70 years old.[4]
The term progeria applies strictly speaking to all diseases
characterized by premature aging symptoms, and is often used as such, and applied
specifically in reference to Hutchinson-Gilford Progeria Syndrome.[2]
It is a rare human
genetic disease linked with a subset of specific mutations within the LMNA gene, coding for
lamin A and lamin C proteins. The LMNA gene is located at position 1q22 and is not usually
inherited, although there is a uniquely inheritable form.[1,5]
LMNA encodes 4 sorts of laminar
proteins, through alternative cutting and splicing,lamin A and C are comparative up to
nucleotide position 574. Lamin A has parent prelamin A,next to post-translational processing,
and generating the mature protein.[3]
In addition to their structural roles they are implicated in
basic nuclear functions such as chromatin organisation, DNA replication, transcription, DNA
repair, and cell cycle progression.[6]
It causes aberrant mRNA splicing, which results in the
assembly of a truncated and partially processed pre-lamin A protein called “progerin”.[7]
Children with Progeria appear normal at birth while the symptoms manifests in first or
second year of life, these patients present stunted growth, muscular atrophy, and skin atrophy,
loss of subcutaneous fat, osteoporosis, arthritis, alopecia, cataracts, diabetes.[1,3]
Death
normally occurs due to complications like atherosclerosis, myocardial infarction, congestive
cardiac failure or coronary thrombosis.[1]
2) EPIDEMOLOGY
HGPS is a lethal, congenital, segemental, premature aging disorder.[7,8,9]
A disease is
considered to be rare(orphan) when it affects one person out of 2000 or less.They are about
5000 to 8000 rare disease most of them are genetic which are chronic disease and are often
fatal .Among these Hutchinson Gilford Progeria is one of the world's rarest disease.[10]
The
estimated prevalence of HGPS is 1 in 20 million people.[11]
The observed male to female ratio](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-2-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1227
of incidence in HGPS is 1.2 :1 and there has been no report on ethnic-specific recurrence.[7]
In 2017 Progeria Research Foundation found that there are 144 cases in 45 countries, of
which 112 children have classic Hutchinson Gilford progeria, and 32 have some progeroid
laminopathy.[3]
In the past 15 years, children with Progeria turn up everywhere around the
world The disease is present in India too.[4]
Generally the disease does not pass from parents
to child as the victim dies before the age of reproduction. It is usually caused by a
replacement (sporadic) mutation during the first division of the cells within the child and
usually genetically dominant; therefore, parents who are healthy will normally not pass it on
to their children.[1]
Death occurs at an average age of 14.6 years.[11]
3) CLINICAL MANIFESTATION
No clinical features present at birth, but within one to two years they begin to display the
effect of accelerated aging means severe growth retardation is usually observed Within the
first year, patients have short stature growth, and weight is more affected than height(median
final height of 100-110cms, median final weight of 10 – 15 kg).[7,12]
Some of the typical physical characteristics of HGPS includes
Alopecia (loss of hair and eyebrows)[7]
Lower weight and loss of subcutaneous fat.[13]
Cranium which appears to be large in comparison with the face.[13]
Thin skin[3,14]
Prominent scalp veins[2,7]
Scleroderma is a transient feature[14]
Micro-ganthia(small jaw)[12]
Narrow face and beaked nose[1]
Protruding ears with absent lobes[13]
Excessive folding on forehead and cheeks[14]
Hip dislocation[4]
Body hair is completely absent[14]
Pyriform thorax.[15]](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-3-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1228
Figure 1: Prominent scalp veins.[16]
4) PHENOTYPES
Osteolysis
There are reports of osteolysis involving the primary ribs.
All these bones are formed by membranous ossification including the center a part of the
distal phalanges.
Osteolysis of the distal phalanges usually starts between 1 and 2 years of age, but can be seen
early as the first months of life or later than 5 years.
The process starts in the index and little fingers, and gradually extends.[14]
Clavicle
The osteolysis starts at the acromial ends of the clavicles, and is merely slowly progressive.At
the early stage it may cause just mild tapering of the distal clavicle.[14]
Skin and hair
Skin changes at the time of birth could also be present. The significant deformity include
glossy and elastic skin. The skin may appear wrinkled with low cutaneous fat.[1]
The loss of subcutaneous fat produces Hypothermia.[3]
The patient is physically weak, when in touch with bright sunlight, hyper pigmentation of
skin may occur with irritation.
Complete loss of hair of all the body parts including scalp, eye lash and skin.[1]](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-4-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1229
Figure 2: Child with Progeria presenting different phenotypes.[17]
5) ETIOLOGY
The only gene associated with HGPS is LMNA gene, in which point mutations are found in
90% of individuals.[13]
HGPS is related to a mutation in ZMPSTE24 that codes for a
metalloproteinase specifically involved in the post-translational proteolytic processing of
prelamin A to mature lamin A, which is responsible for scaffolding and organizing the
nuclear envelope surface.[15]
6) GENERAL MECHANISM OF LAMIN REGULATED EXPRESSION
HGPS belongs to a set of disorders called laminopathies which affect nuclear lamins and
build up a number of mutations within the gene LMNA, which are recognizised within the
major cases of HGPS. The gene LMNA encodes nuclear lamin A, with the predominant
vegetative cell isoforms lamin A and C arising by alternative RNA splicing, which underlies
and organizes the inner surface of the nuclear envelope. There are at least 11 distinct diseases
associated with >300 different mutations in LMNA.[15]
The most overt cellular defects in
HGPS are dramatic changes in nuclear morphology, a phenotype that is not surprising given
the prominent architectural role of lamin A in the nucleus. Whereas the lamin proteins in
healthy cells move dynamically between the nuclear lamina polymer at the nuclear periphery
and the nucleoplasm, they become immobilized in HGPS patient cells, leading to thickening
of the lamina.[18]
The lamins belong to the multiprotein-family of intermediate filaments, and](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-5-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1230
consist of an N-terminal head domain, an alpha-helical (coiled-coil) rod domain important for
the dimerization, and a usually globular C-terminal tail domain. Lamins are located in the
nuclei at position 1q22 of multicellular eukaryocytes.[5,14]
There are two major types of lamins: the B-type lamins, indispensable for replication and
transcription and expressed in all cells (cells are not viable without Lamin B) and the A-type
lamins, expressed in all differentiated cells. LMNA encodes four A-type lamin isoforms:
Lamin A, AD10, C, and C2, generated by alternative mRNA splicing.[14]
They have many
functions: they provide nuclear envelope its mechanical strength which determine the nuclear
shape, nuclear pore complexes, and form the structure in which many other proteins anchor.
They can be regarded as the main determinants of the nuclear architecture. In addition, lamins
are essential for DNA replication and mRNA transcription and have functions in gene
regulation and many signal transduction pathways, both by themselves has direct interactions
with the DNA or a wide range of protein partners, chromatin organization and cell cycle
progression.[6,14,19,20]
Lamin A and Lamin C are the two abundant structural proteins of the nuclear lamina which
are the products of an equivalent gene, LMNA. Lamin A is twelve exon protein. Prelamin A,
the precursor of Lamin A, involves the splicing from middle of exon 10 to exon 11 then to
exon 12.[1]
Prelamin A has CAAX (where the C is a cysteine, the A residues are aliphatic
amino acids), and the X can be any amino acids.[7,12,15]
This terminal triggers farnesylation of
the carboxy terminal cysteine (the C of the CAAX tetrapeptide) by a cytosolic enzyme,
known as protein farnesyl transferase.[6,21]
The farnesylated Prelamin A attaches with the
Endoplasmic Reticulum.[1]
The farnesylation increases lipophilicity of lamin-A and increases its membrane
association.[12]
Following farnesylation, the ending three amino acids of Prelamin A are
separated by an Endoprotease.[1]
The enzymes accountable for liberation of these amino acids
are: a Zinc metalloproteinase ZMPSTE24[12]
and a prenyl protein endopeptidase RCE1.[1]
After the liberation of the terminal amino acids, farnesyl-cystein residue is methylated by an
enzyme Isoprenylcystein Carboxy Methyl Transferase (ICMT).[22]
In the final step of Prelamin A including farnesyl cysteine methyl ester are released off by
ZMPSTE24 and mature Lamin A is released from endoplasmic reticulum into cytosol. The](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-6-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1231
emerging protein, now Lamin A is not any more membrane-bound, and carries out the
outcomes inside the nucleus.[1,12,22]
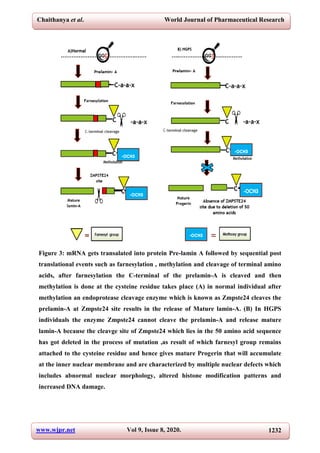
7) MUTATION IN LAMIN-A CAUSES PROGERIA
The genetic basis for HGPS was not found until it had been found to be autosomal-dominant
which are caused by a repeated, dominant, de novo heterozygous gene mutation in position
1824 of the LMNA gene, replacing cytosine with thymine. Progerin, of the prelamin A
protein whose further processing is abnormal.[2,7]
The mutation G608G of HGPS and
therefore the consequent abnormal splicing produce a prelamin A that also retains the CAAX
box, but is missing a neighbourhood for endoproteolytic cleavage.[7,15]
One mutation in the
gene LMNA that causes HGPS consists of the de novo substitution of exon 11 of LMNA
(c.1824C>T), which produces activation of a cryptic splice site.[3,23,24]
This mutation causes
aberrant splicing in exon 11 and the deletion of 50 residues close to the C terminus of lamin-
A, including the second ZMPSTE24 cleavage site.This deletion prevents complete processing
of prelamin-A, resulting in the accumulation of a farnesylated lamin A,known as
progerin.[3,15]
Progerin, unlike mature lamin-A, persist farnesylated, it gains lipophilicity with
the nuclear membrane, therefore causing an obtrusion within the integrity of the nuclear
lamina. Indeed, HGPS patient cells show variety of abnormalities in nuclear structure and
performance. Upon fluorescence microscopy labelling with antibodies directed against lamins
A/C, fibroblasts from individuals with HGPS were characterised by the presence of
dysmorphic nuclei with altered size and shape, presence of lobules, wrinkles, herniations of
the nuclear envelope, thickening of the nuclear lamina, loss of peripheral heterochromatin,
and clustering of nuclear pores.[3,7,15,22]
Small amount of progerin is extremely potent in terms
of causing disease phenotypes in humans and in causing misshaped nuclei in cultured
cells. Supporting the theory that progerin endeavour dominant negative impact HGPS.[7]](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-7-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1233
8) DIAGNOSIS
To diagnose progeria, doctors observed phenotypes like symptoms such as, skin changes and
a failure to gain weight, and as well as x-rays of patients and on the basis of the excretion of
the glycosaminoglycan and urinary hyaluronic acid testing and radiography.[12]
a) Urinary hyaluronic acid test
The most frequent sign of HGPS is elevated levels of hyaluronic acid excretion in the urine of
patients with HGPS, but are not diagnostic. After performing the urinary hyaluronic test on
HGPS patient it was found that hyaluronic acid level elevated in urine and decreased level of
primary antioxidant enzymes in the blood as well as certain fatty acid compound. Due to
decreased level of antioxidant enzymes in the blood, it may cause aging which believed to be
a buildup oxidant in the blood.[3,12,15]
Hyaluronic acid is a non-sulfated glycosaminoglycan
maintaining skeletal, muscular, cutaneous, and vascular integrity and texture. These
hyaluronic acid abnormalities may account for hardened collagen, calcification of the arterial
walls, and changes in the skin which look very much like scleroderma.[3]
b) Genetic screening
On a genetic level, the screening of correlated mutations is performed by sequencing of
LMNA gene. The diagnosis for HGPS is clinically performed with the subsequent screening
of gene mutation LMNA, which uncovers point mutations in the patients with HGPS, and the
test for uniparental disomy of chromosome 1 and deletions associated with HGPS.[3,15]
Discovery of mutant lamin A gene, nowadays helpful for detection of the elevated mutant
gene, that mutant gene identify from a blood sample and skin biopsy of the patient, this gives
definite diagnosis report.[12,16]
c) Prenatal testing
Investigation of DNA extraction obtained by aminocentesis from the fetal cell of HGPS
children applied for prebirth diagnosis, mostly performed on about 15-18 weeks gestation.[12]
d) Radiological diagnosis
Radiological finding of the common LMNA truncating mutation could also be helpful in the
diagnosis. The characteristic radiological abnormalities are to be found in the skull, thoracic
cage, long bones.](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-9-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1234
The cranial bones tend to be hypoplastic, fontanels and sutures remain open longer than
expected.The progressive bone loss from the distal phalanges of the fingers and toes is one
hallmarks of the disease.[12,13]
e) Laboratory studies
There is low levels of blood cholesterol are limited to low high-density lipoprotein levels.[12]
Testing can help in the clinical findings of progeria and to evaluate for the potential for
increasing atherosclerosis during early childhood.
Testing might include a HDL blood test, which can reveal a low level of high-density
lipoprotein (HDL) cholesterol, the "good" cholesterol that helps keep arteries open.[2]
9) HGPS AND NORMAL AGING
The similarities between HGPS and normal aging extend to all levels, from shared molecular
features to similarities in symptoms.
Notably, progerin is produced in normally aged individuals as well as in HGPS patients. This
is because the classical HGPS mutation is a pre-mRNA splicing mutation.[18]
Cells from healthy aged individuals also express low levels of progerin, resulting in similar
phenotypes.
For instance, the levels of γH2AX, DNA DSBs and abnormal nuclei increase with an
individual’s age. Induced DSBs in the same cells can be repaired efficiently.[23]
Preliminary studies indicate that the level of chromatin-bound XPA is much higher in older
HGPS cells. Interestingly, chromatin-bound XPA also was higher in the cells from normal
older individuals than in cells from younger individuals.
Thus, HGPS or related laminopathies are an excellent model for the study of normal human
aging.[23]
Notably, the atherosclerotic plaques in HGPS are similar to those found in aging
individuals. Additionally, vascular stiffening in progeria is much like that seen over a lifetime
of aging reflected in each population by increased pulse wave velocity.
HGPS is a primary vasculopathy, characterized by early and pervasive accelerated vascular
stiffening followed by hypertension, vessel plaques, angina, cardiomegaly, metabolic](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-10-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1235
syndrome, and congestive heart failure. Isolated from risk factors such as
hypercholesterolemia and increased C-reactive protein.[18]
10) TREATMENT
Presently, HGPS has no cure, and there has been no success in finding a specific treatment.
However, some options exist that attempt to increase the quality of life in patients.[3]
a) Farnesyl transferase Inhibtors – Lonafarnib
As progerin is permanently farnesylated, researchers initially turned to farnesyltransferase
inhibitors (FTIs) within the look for a pathogenic treatment.
The medication will also get involved in the farnesylation of lamin B1 and B2, perhaps
causing more deface to the nuclear lamina. Finally, there was a concern that prelamin-A
might be geranylgeranylated in the presence of FTI.[7]
The farnesyl group is synthesized through the cholesterol biosynthetic pathway, and
medicines like statins and bisphosphonates are known to scale back its production.
Lonafarnib will be taken orally, twice per day. Every patient will start Lonafarnib therapy at a
dose of 115mg/kg and was escalated to 150mg/kg. FTI treatment caused defects in
centrosome separation leading to donut-shaped nuclei.[4,7,20,22]
b) Statins and Bisphosphonates
Statins and aminobisphosphonates, both inhibit protein prenylation than FTIs and alter
cholesterol biosynthetic pathway.[7]
Pravastatin is traded as Pravachol or Selektine and it is included in the family of statins. As
well as zelodronate (also mentioned as Zometa and Reclast, which is a bisphosphonate), its
utility in Hutchinson-Gilford Progeria Syndrome (HGPS) is that the prevention of farnesyl
groups formation, which leads to cause of disease.[2]
Zoledronic acid is a bisphosphonate, used to reduce osteoporosis.[1,22]
c) Rapamycin
The effect of rapamycin is inhibition of mammalian target of rapamycin (mTOR) pathway by
rapamycin.[7]](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-11-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1236
Rapamycin, caused removal of progerin from the nuclear membrane through autophagy.[2]
Rapamycin, also referred to as Sirolimus, may be a macrolide.
There are recent studies concerning rapamycin which conclude that it can minimize the
phenotypic effects of progeria fibroblasts.[2,4,7]
d) RNA therapy
Because the HGPS mutation results from the activation of an alternative pre-mRNA splice
site, HGPS may be a prime candidate for an RNA therapy approach via inhibition of this
site.[18]
They used antisense morpholino oligonucleotides specifically directed against the aberrant
exon 11 and exon 12 junction contained in mutated pre-mRNAs to focus the splicing defect
observed in HGPS, and consequently decrease the production of progerin.[7]
This strategy was effectively tested to a knockin HGPS mouse model resulting in improved
body weight, increased life expectancy and rectifying mutant phenotypes and an additional
approach is to get rid of progerin mRNA using siRNA based methods.[18]
e) Gene therapy
As the progerin protein acts in a dominant fashion and its effects cannot be compensated by
introduction of wild-type lamin A, gene therapy for HGPS focuses on targeted gene
correction.
For example, zinc-finger, TALEN, or CRISPR-based approaches, in which the LMNA is
repaired ex vivo and corrected cells are re-introduced into patients, is a possibility, albeit a
technically challenging one.[18,22]
As proof of principle for the possibility of this perspective,rectifying of the genetic defect in
HGPS patient derived iPS cells and their following differentiation has been accomplished.[18]
f) Stem cell treatment
In vitro, progerin affects the multipotency and differentiation of human mesenchymal stem
cells and HGPS patient-derived iPS cells exhibit differentiation defects.[18]](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-12-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1237
Figure 4: Drugs that involve in inhibiting the production of farnesyl group, act on the
biosynthetic pathway of cholesterol (statins and bisphosphonates). Farnesyl tranferase
inhibitors(FTI’s) mainly inhibit the enzyme farnesyl transferase and prevent the
farnesylation reaction on Prelamin-A.
11) SUPPORTIVE THERAPY
Treatment is aimed at controlling the devastating effects caused by premature aging.
Prescribed formulas of antioxidants, vitamins, lipoic acid, and coenzyme-Q are used to
increase antioxidant levels. Low antioxidant levels are common in persons with progeria and
cause cellular destruction.[2]
1) Aspirin: Aspirin is now accepted as a crucial weapon in the prevention of heart condition.
Recent clinical trials have shown that aspirin reduces the danger of transient ischemic attacks
(TIA) strokes and heart attacks by inhibiting platelet aggregation.](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-13-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1238
Dosage is fixed based on patient weight, and should be given 2-3mg/kg once daily or
alternate day. This is of benefit to people with narrowed coronary arteries which is common
place in children with Progeria.[1,4,12,25]
2) Hydrotherapy: Hydrotherapy results in relaxation, pain reduction and assists mobility. It
also can help prevent arthritis from getting worse.[1,4,25]
3) Vitamin E: Vitamin E is a fat-soluble vitamin that protects Vitamin A and essential
fattyacids from oxidation in the body cells and prevents breakdown of body tissues.[4]
Antioxidants such as Vitamin E act to ensure the cells against the harmful effects of
free radicals.[1,4]
Free radicals will damage the cells and eventually leading to cardiovascular
disease.[1,4,25]
4) Fluoride: All Progeria children have problems with their teeth, under development of the
facial bones and the lower jaw leads to delayed eruption of the teeth, they can be small,
irregularly formed or even missing and tooth decay is common. Fluoride can greatly help
dental health by strengthening the tooth enamel, making it more resistant to tooth decay.[1,4,25]
5) Nutrini: Patients have a very small appetite and don’t really enjoy eating. Nutrini provides
all the nutrients essential for well-being[1,4,25]
and it is a sole source of which contain source
Sodium caseinate
Maltodextrin
Beta-carotene
Vegetable oils (rapeseed oil,sunflower oil) Fish oil,
Cyanocobalamin
Phytomenadione
D-biotin
Calcium D-pantpthenate
Ferrous lactate
Sodium chloride
Potassium chloride and calcium hydroxide.
(https://www.nutriciahcp.com/uploadedFiles/Main/Sub_sites/ONS_Site/ons/shop/Datacard_0
7022019_Nutrini(2).pdf](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-14-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1239
6) Pro-Cal
Pro-Cal may be a new generation protein and calorie food which will be added to a good sort
of food and drink to complement the energy and protein content of the normal diet with the
minimum effect on taste, volume and texture.[1,4,25]
CONCLUSION
Since HGPS is a rare disease and affects only a small percentage of the population around the
world, the support and resources for the invention of newer sites of disease pathology and
treatment options are less. Progress in the HGPS field is increasing over the past decade
cardiovascular failure, myocardial infarction, stroke are the remarkable causes of death ,
notebly these dysfunctions in cardiovascular system are similar to those found in aging
persons, this aspects of it has gained a lot of attention to study the normal aging process.
Therapies like Gene therapy, RNA therapy and stem cell therapy are quiet promising in the
future times.
Source of funding: None.
Conflict of interest: None.
REFERENCES
1. Rakha P, Gupta A, Dhingra G, Nagpal M. Hutchinson-Gilford progeria syndrome: a
review. Der Pharmacia Sinica, 2011; 2(1): 110-7.
2. Neelam S, Krishna M, Semwal BC, Shravan P, Kuldeep S, Deepak S. Progeria: A
Review. Int J Pharm Sci Rev Res., 2012; 14(1): 4449.
3. Camacho-Cruz J, Dary Gutiérrez-Castañeda L, Pulido D, Echeverri C, Bernal B, Bautista
L, et al. Hutchinson-Gilford Progeria Syndrome. Int J Pediatric, 2019; 7(10): 10283-9.
4. Kumar P. An Overview: Projeria. Pharmacologyonline, 2011; 3: 176-84.
5. Piekarowicz K, Machowska M, Dzianisava V, Rzepecki R. Hutchinson-Gilford progeria
syndrome current status and prospects for gene therapy treatment. Cells, 2019; 8(2): 88.
6. Prokocimer M, Barkan R, Gruenbaum Y. Hutchinson–Gilford progeria syndrome through
the lens of transcription. Aging Cell., 2013; 12(4): 533-43.
7. Baek JH, McKenna T, Eriksson M. Hutchinson-gilford progeria syndrome. Genetic
Disorders, 2013; 1: 65-87.](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-15-320.jpg)
![Chaithanya et al. World Journal of Pharmaceutical Research
www.wjpr.net Vol 9, Issue 8, 2020. 1240
8. Gerhard-Herman M, Smoot LB, Wake N, Kieran MW, Kleinman ME, Miller DT, et al.
Mechanisms of premature vascular aging in children with Hutchinson-Gilford progeria
syndrome. Hypertension, 2012; 59(1): 92-7.
9. Rodriguez S, Coppedè F, Sagelius H, Eriksson M. Increased expression of the
Hutchinson–Gilford progeria syndrome truncated lamin A transcript during cell aging.
Eur J Hum Genet, 2009; 17(7): 928-37.
10. De Vrueh R, Baekelandt ER, De Haan JM. Background paper 6.19 rare diseases. World
Health Organization, Geneva, 2013 Mar 12.
11. Hamczyk MR, Villa‐Bellosta R, Quesada V, Gonzalo P, Vidak S, Nevado RM, et al.
Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular
smooth muscle cells. EMBO Mol Med., 2019; 11(4): 9736.
12. Devi S, Singh K. Risk factors, prevalence and diagnosis of hutchison gilford syndrome
with special reference to case reports. Int J Pharm Pharm Sci., 2003; 9: 1-5.
13. Faivre L, Cormier-Daire V. Progeria. Orphanet encyclopedia, 2005.
14. Hennekam RC. Hutchinson–Gilford progeria syndrome: review of the phenotype. Am Jo
Med Genet Part A., 2006; 140(23): 2603-24.
15. Coutinho HD, Falcão-Silva VS, Gonçalves GF, da Nóbrega RB. Molecular ageing in
progeroid syndromes: Hutchinson-Gilford progeria syndrome as a model. Immun Ageing,
2009; 6(1): 1-7.
16. Gordon LB, Brown WT, Collins FS. Hutchinson-Gilford progeria syndrome.
InGeneReviews®[Internet] 2019 . University of Washington, Seattle.
17. Devi AS, Thokchom S, Devi AM. Children Living with Progeria. Nurse Care Open
Acces J, 2017; 3(4): 77.
18. Gordon LB, Rothman FG, Lopez-Otin C, Misteli T. Progeria: a paradigm for translational
medicine. Cell, 2014; 156(3): 400-7.
19. Datta S, Snow CJ, Paschal BM. A pathway linking oxidative stress and the Ran GTPase
system in progeria. Molecular Bio Cell, 2014; 25(8): 1202-15.
20. Gordon LB, Kleinman ME, Massaro J, D’Agostino Sr RB, Shappell H, Gerhard-Herman
M, et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and
zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation,
2016; 134(2): 114-25.
21. Cao K, Capell BC, Erdos MR, Djabali K, Collins FS. A lamin A protein isoform
overexpressed in Hutchinson–Gilford progeria syndrome interferes with mitosis in
progeria and normal cells. Proc Natl Acad Sci., 2007; 104(12): 4949-54.](https://image.slidesharecdn.com/progeria-200926031351/85/Progeria-16-320.jpg)








![[Speech3] Draft002](https://cdn.slidesharecdn.com/ss_thumbnails/63571e48-dcb3-4e06-bde3-3a90af7bd448-160809163414-thumbnail.jpg?width=640&height=640&fit=bounds)



























































