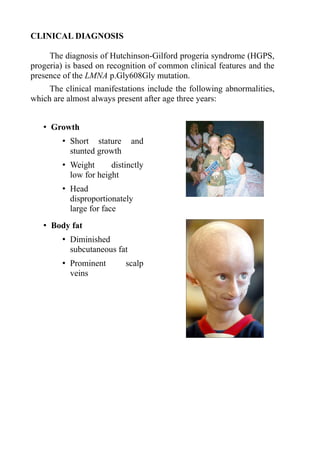
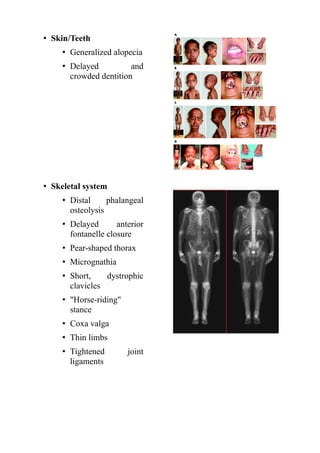
This document provides information about Progeria Syndrome, a rare genetic condition causing rapid aging in children. Key points:
- Progeria is caused by a dominant mutation that prevents normal telomere lengthening, leading to premature aging symptoms. It affects 1 in 8 million live births.
- Symptoms start in the first year of life and include failure to thrive, hair loss, stiffness, small head and body size. Life expectancy is around 13 years due to heart disease or stroke.
- Diagnosis is based on clinical features and detecting the recurrent LMNA gene mutation found in all Progeria cases. Prenatal testing is possible through amniocentesis.







![TESTING:
The clinical diagnosis of HGPS is based on recognition of
common clinical features and detection of the p.Gly608Gly mutation
in exon 11 of the LMNA gene, which is present in all individuals with
HGPS. Molecular genetic testing for this mutation is clinically
available.
Urinary hyaluronic acid. Although urinary hyaluronic acid has
been reported to be increased in most children with HGPS [Brown et
al 1990], the measurement is now regarded as unreliable [Gordon et al
2003] and is not recommended for diagnosis.
Molecular Genetic Testing
GeneReviews designates a molecular genetic test as clinically
available only if the test is listed in the GeneTests Laboratory
Directory by either a US CLIA-licensed laboratory or a non-US
clinical laboratory. GeneTests does not verify laboratory-submitted
information or warrant any aspect of a laboratory's licensure or
performance. Clinicians must communicate directly with the
laboratories to verify information.](https://image.slidesharecdn.com/progeriaproject-100317044956-phpapp02/85/Progeria-Syndrome-10-320.jpg)






![RISK TO FAMILY MEMBERS
Parents of a proband
• All probands with HGPS have the disorder as the result of a de
novo mutation.
• Parents of probands are not affected.
Sibs of a proband
• Because HGPS is caused by a de novo mutation, the risk to the
sibs of a proband is small.
• One instance of apparent somatic and germline mosaicism has
been reported [Wuyts et al 2005]. Therefore, the recurrence risk
may be on the order of one in 500, as in other de novo dominant
mutations.
• With the exception of two sets of identical twins with HGPS, the
authors are unaware of any convincing cases of a family with
more than one sib with classic HGPS.
Offspring of a proband. Individuals with HGPS do not reproduce.
Other family members of a proband. Because HGPS occurs as the
result of a de novo mutation, other family members of a proband are
not at increased risk.](https://image.slidesharecdn.com/progeriaproject-100317044956-phpapp02/85/Progeria-Syndrome-17-320.jpg)
![GENETICALLY RELATED (ALLELIC) DISORDERS
More than ten other diseases and conditions with mutations or
variations in the LMNA gene have been identified. See OMIM
150330.
Progeroid laminopathy. The term "progeroid laminopathy" can be
used to describe phenotypes that resemble HGPS in which an LMNA
mutation other than p.Gly608Gly has been identified.
Approximately 10% of individuals with
clinically diagnosed HGPS have
uniparental isodisomy (UPD) of
chromosome 1 (including the LMNA
gene), a mosaic rearrangement of
chromosome 1, and a deletion involving
the LMNA gene locus [Eriksson et al
2003]. These individuals are
hypothesized to have somatic changes
that occur in vivo or in vitro, deleting the
mutated LMNA and providing a growth
advantage to that clone of cells.
Penetrance: Penetrance is complete.
Differential Diagnosis
The following are other syndromes that include some features of
premature aging:
• Neonatal progeroid syndrome (Weidemann-Rautenstrauch
syndrome)
• Acrogeria
• allermann-Streif syndrome
• Gerodermia osteodysplastica
• Berardinelli-Seip lipodystrophy ( generalized lipodystrophy)
• Petty-Laxova-Weidemann progeroid syndrome
• Ehlers-Danlos syndrome, progeroid form
• Werner syndrome.](https://image.slidesharecdn.com/progeriaproject-100317044956-phpapp02/85/Progeria-Syndrome-18-320.jpg)





























































![[Speech3] Draft002](https://cdn.slidesharecdn.com/ss_thumbnails/63571e48-dcb3-4e06-bde3-3a90af7bd448-160809163414-thumbnail.jpg?width=640&height=640&fit=bounds)

































