Downloaded 170 times






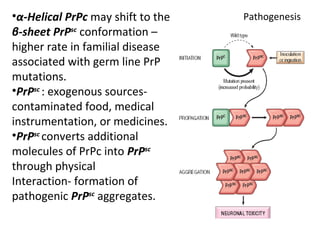



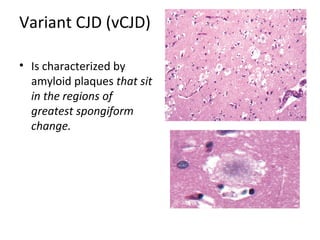








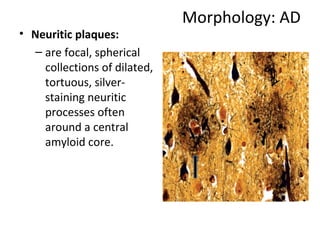
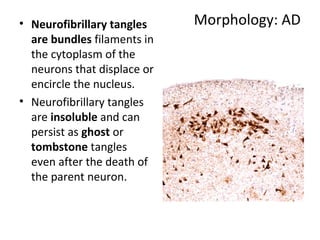
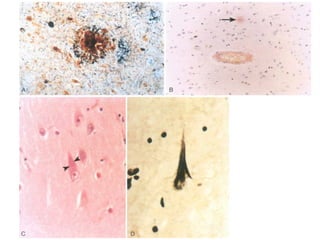




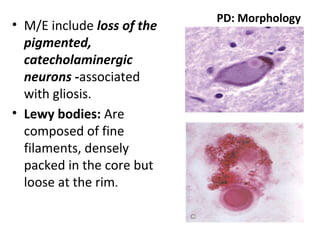




This document discusses several neurodegenerative diseases including prion diseases, Alzheimer's disease, and Parkinson's disease. Prion diseases are a group of rare progressive conditions caused by prions, which are abnormal folded proteins. Common types include Creutzfeldt-Jakob disease and variant CJD. Alzheimer's disease is characterized by beta-amyloid plaques and neurofibrillary tangles leading to dementia. Parkinson's disease results from degeneration of dopaminergic neurons and is associated with Lewy bodies containing alpha-synuclein.






















































































