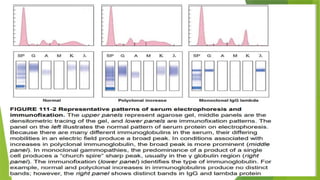
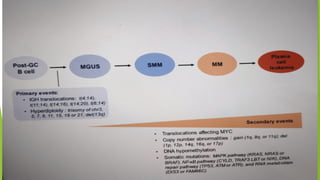
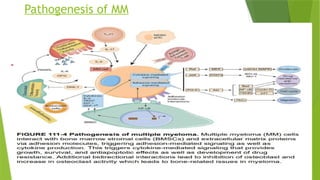
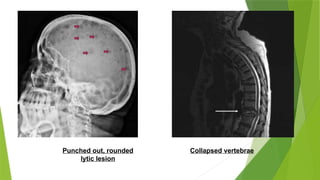
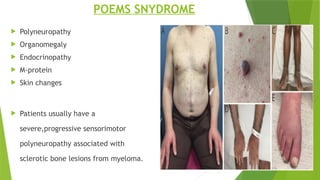
This document discusses plasma cell disorders, primarily focusing on multiple myeloma, Waldenstrom's macroglobulinemia, and related conditions, outlining their definitions, epidemiology, risk factors, clinical features, diagnosis, and treatment strategies. It highlights the malignant proliferation of plasma cells, the associated clinical manifestations, and the specific management approaches for different stages and symptoms of these disorders. The treatment recommendations include systemic therapy, high-dose therapy with autologous stem cell transplantation, and supportive care measures tailored to each patient's needs.

























































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)

















