Downloaded 188 times




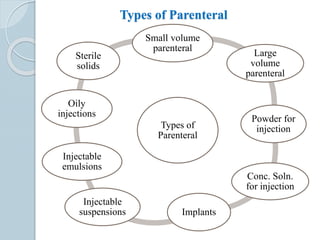
















































































































The document discusses parenteral formulations, which are sterile dosage forms administered directly into the body rather than orally. It defines parenterals and describes various types including small and large volume injections, powders, and implants. Key factors in parenteral design are discussed such as drug solubility, vehicle selection, dosage form, ingredients, pH, and color. Common vehicles like water and buffers are outlined. The document also covers routes of administration, isotonicity, production facilities and procedures, and quality control testing for parenteral products.






















































































