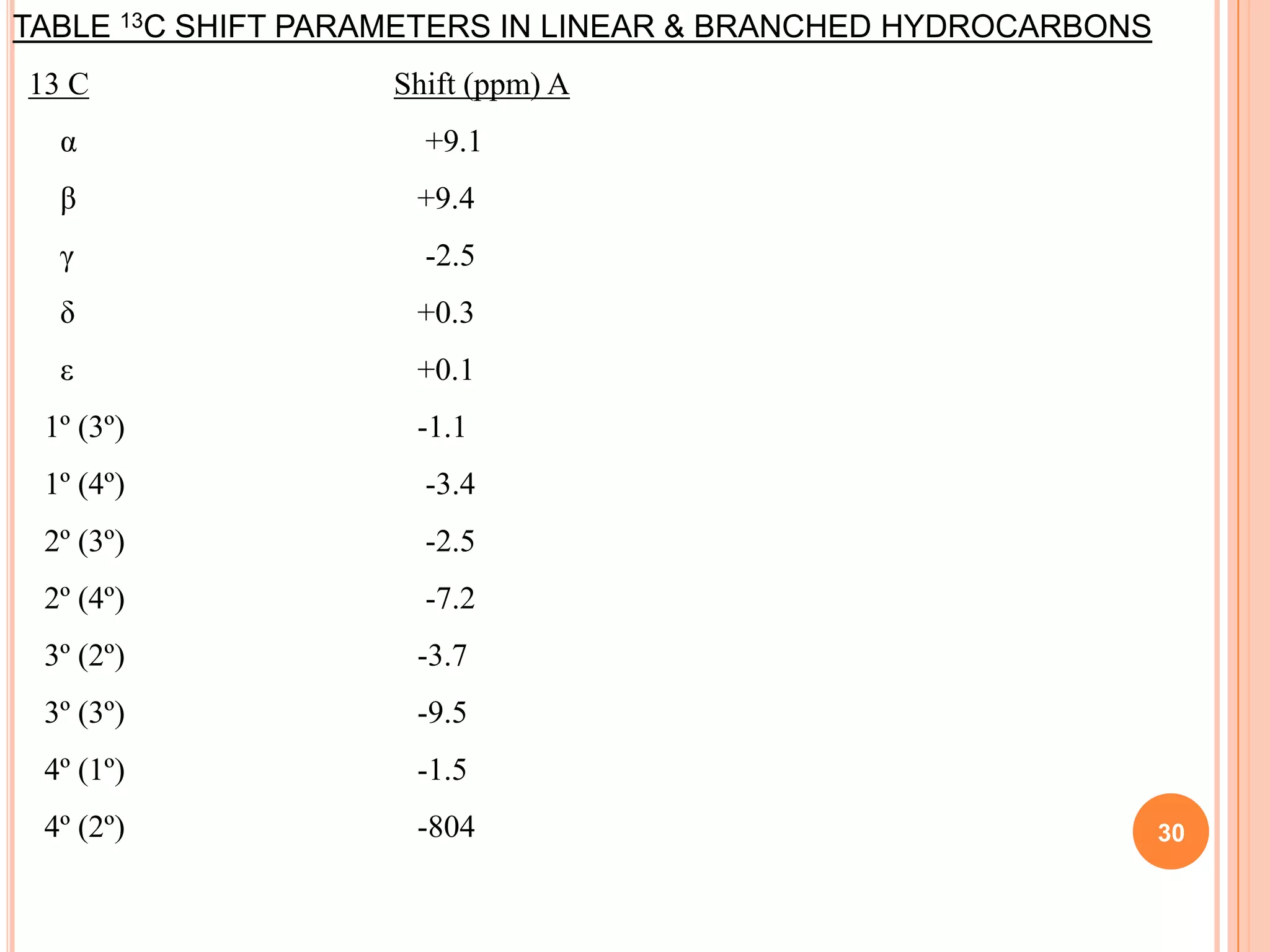
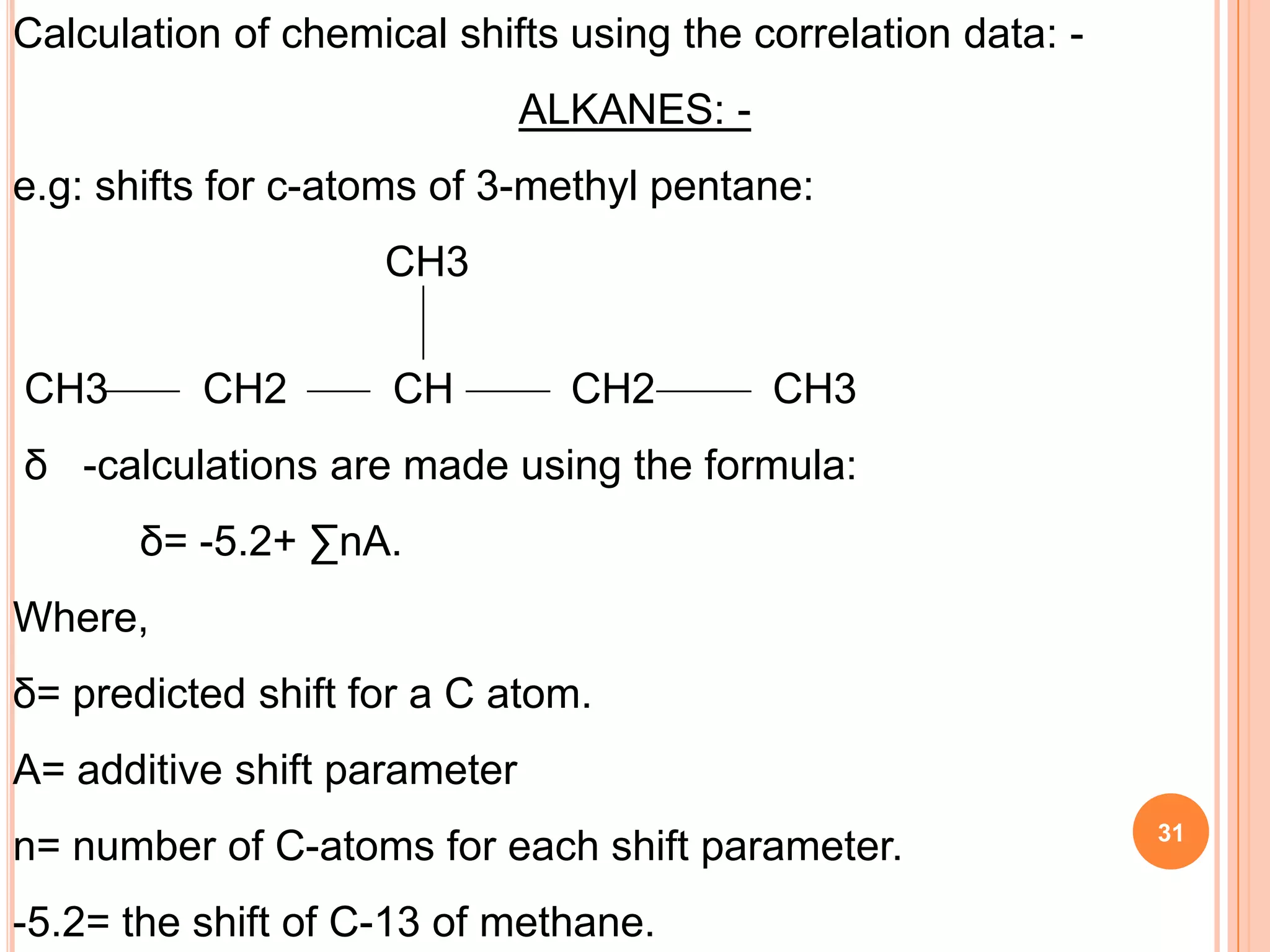
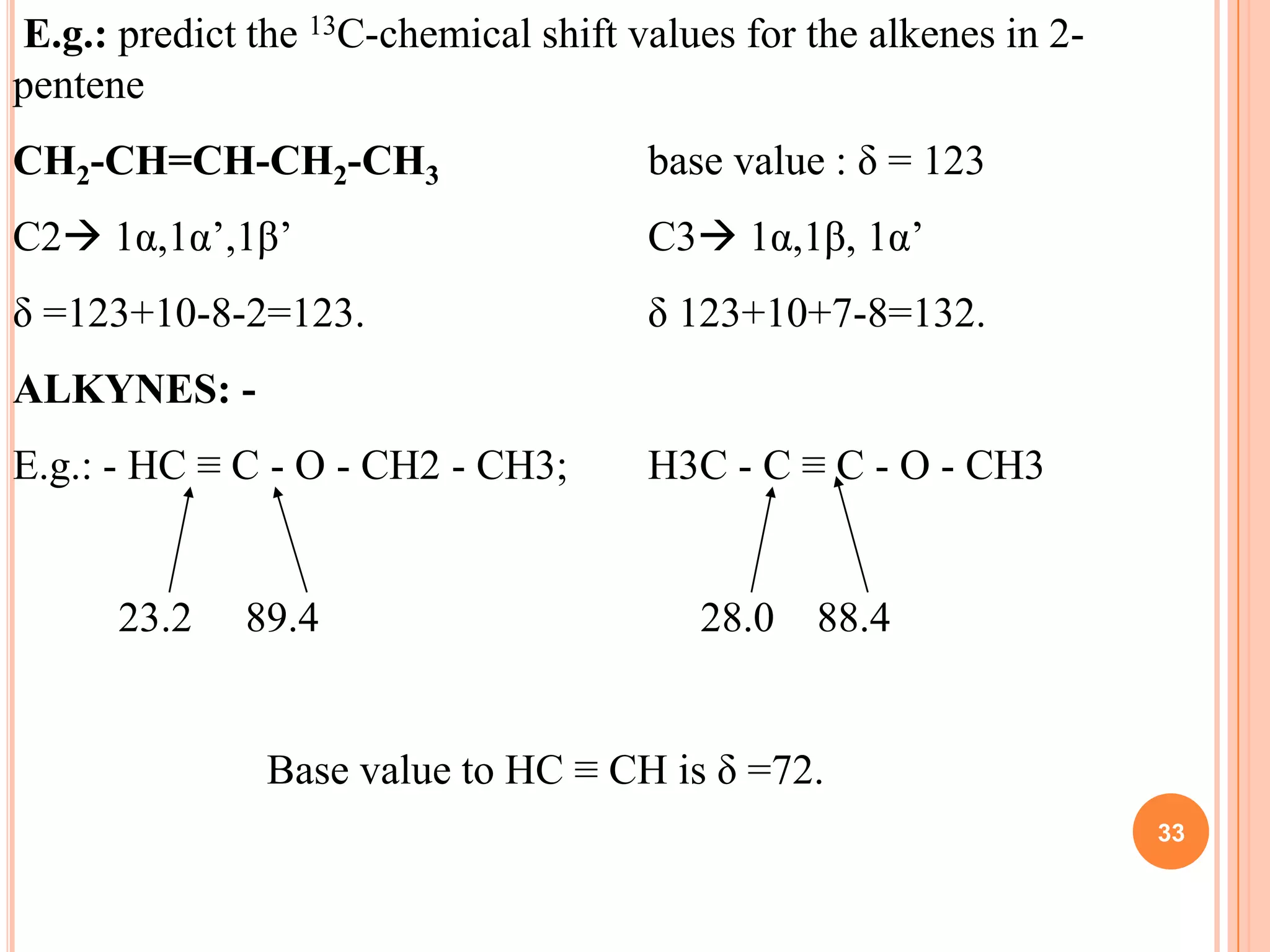
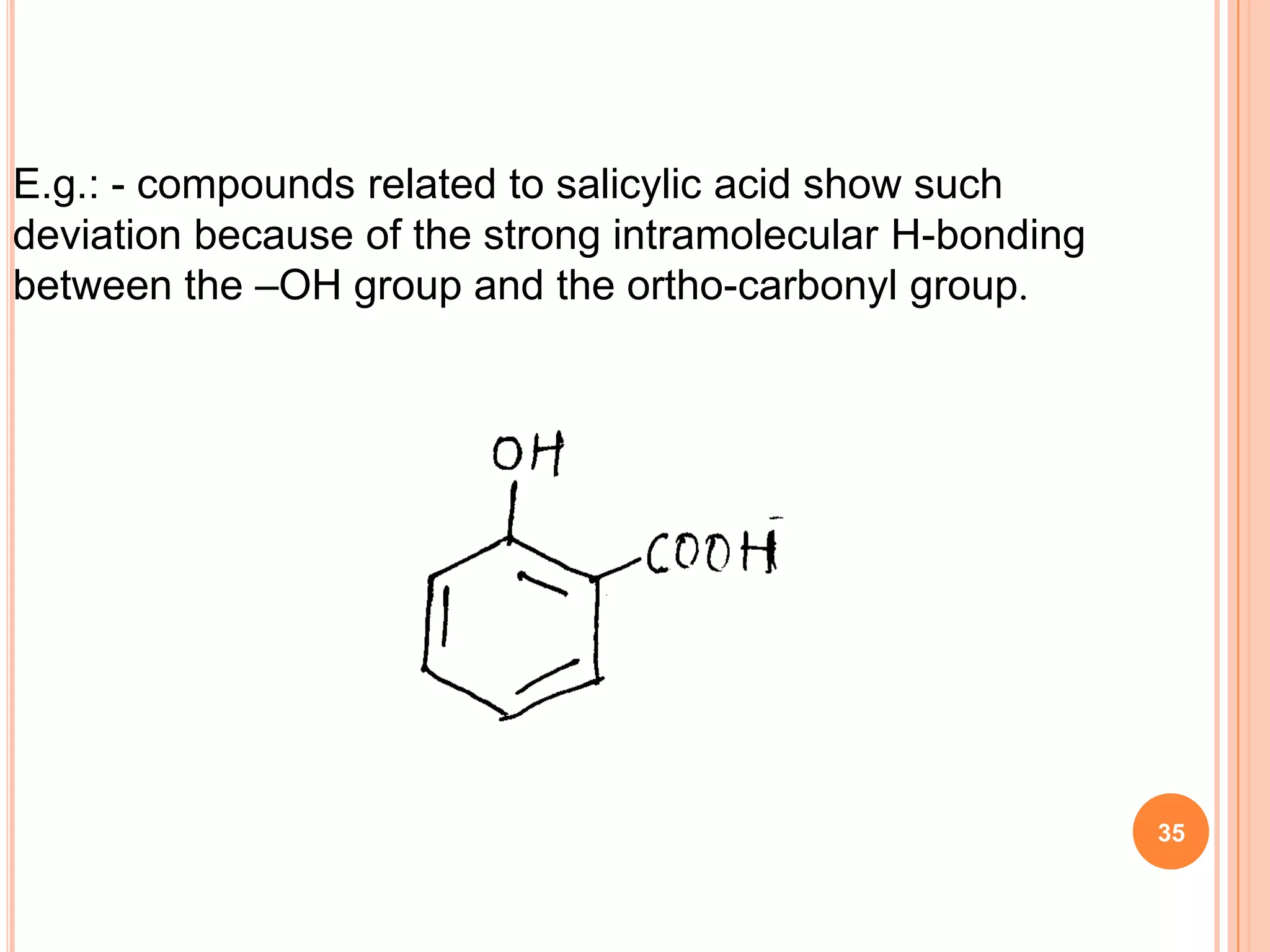
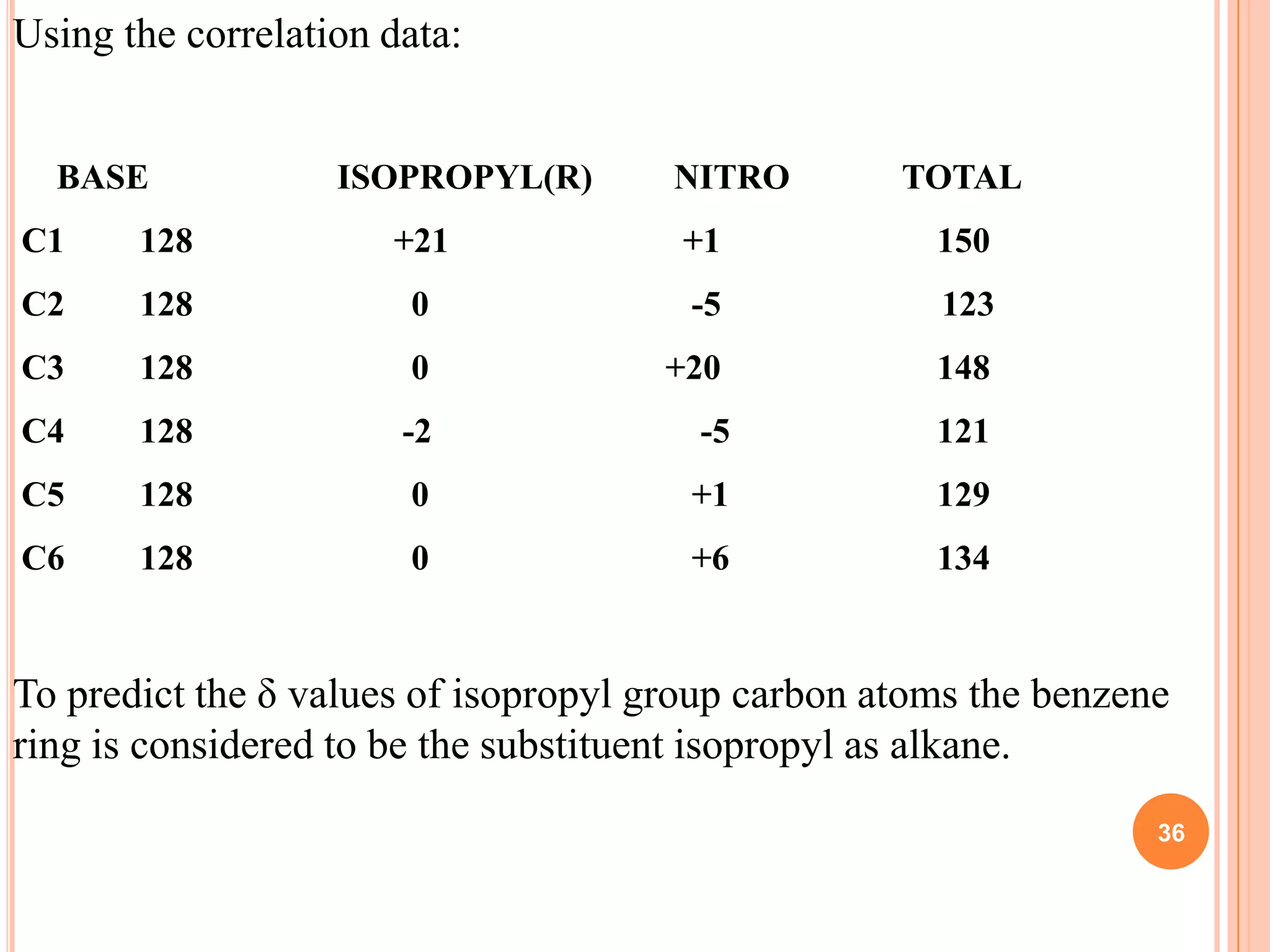
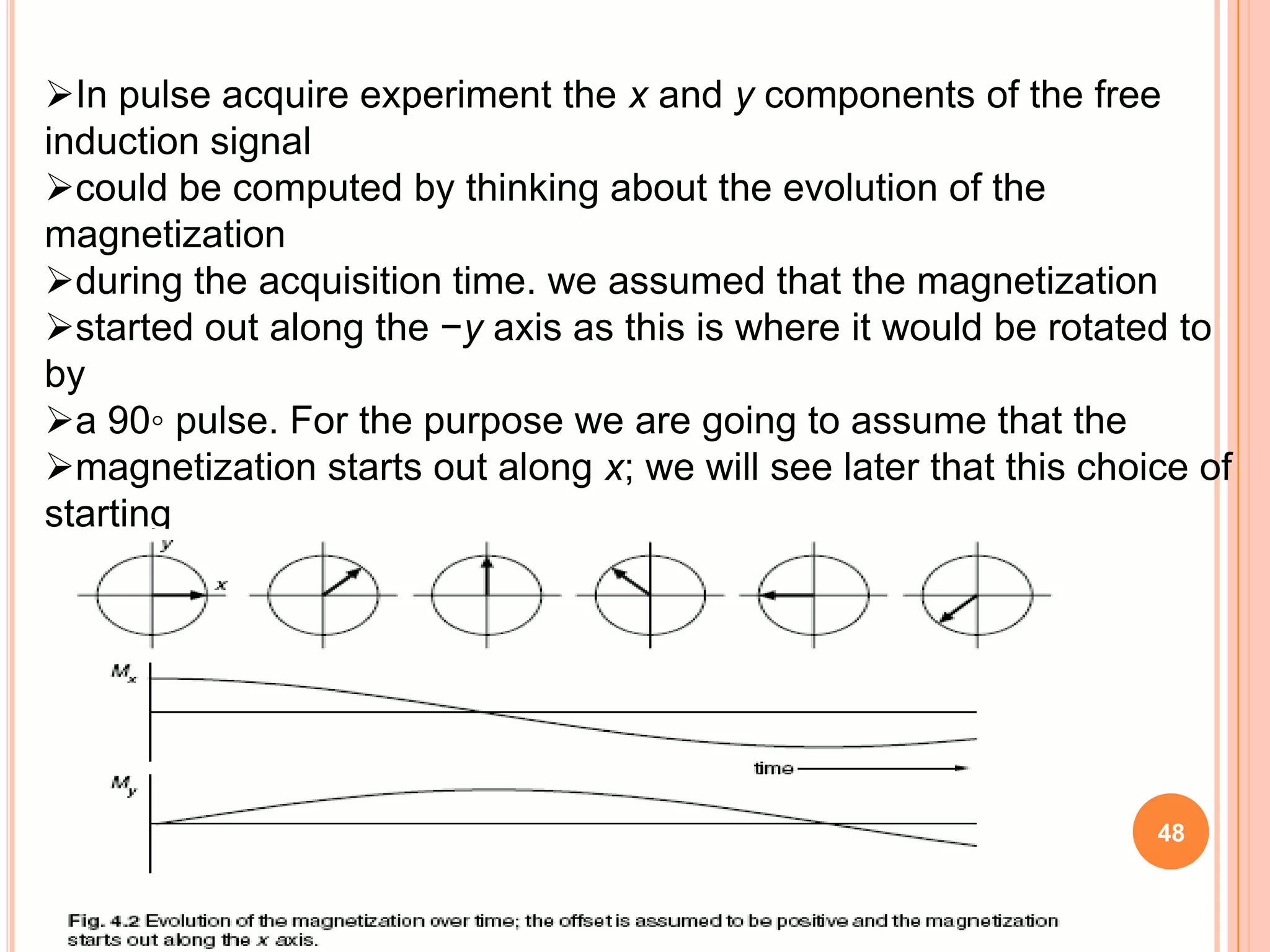
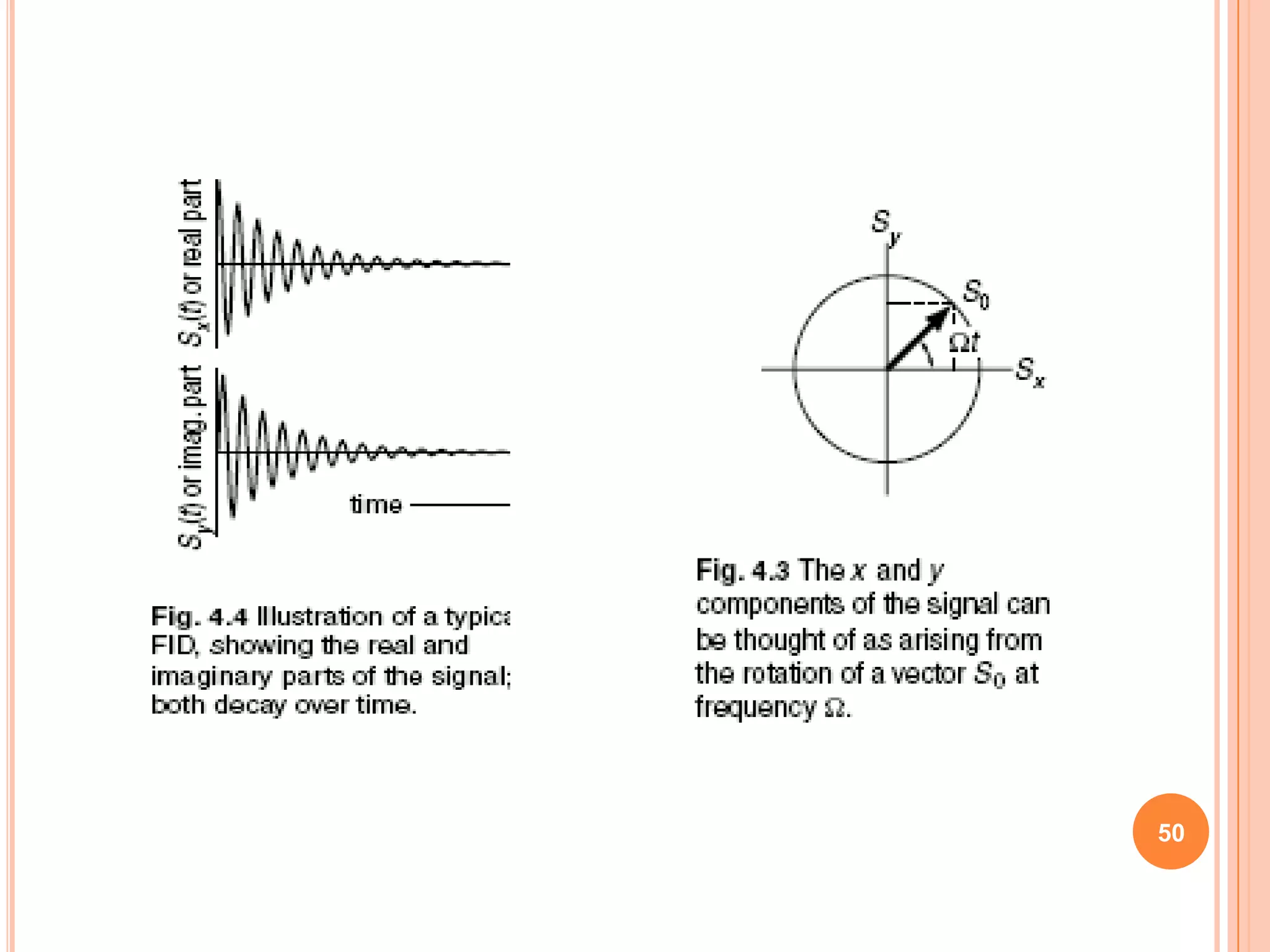
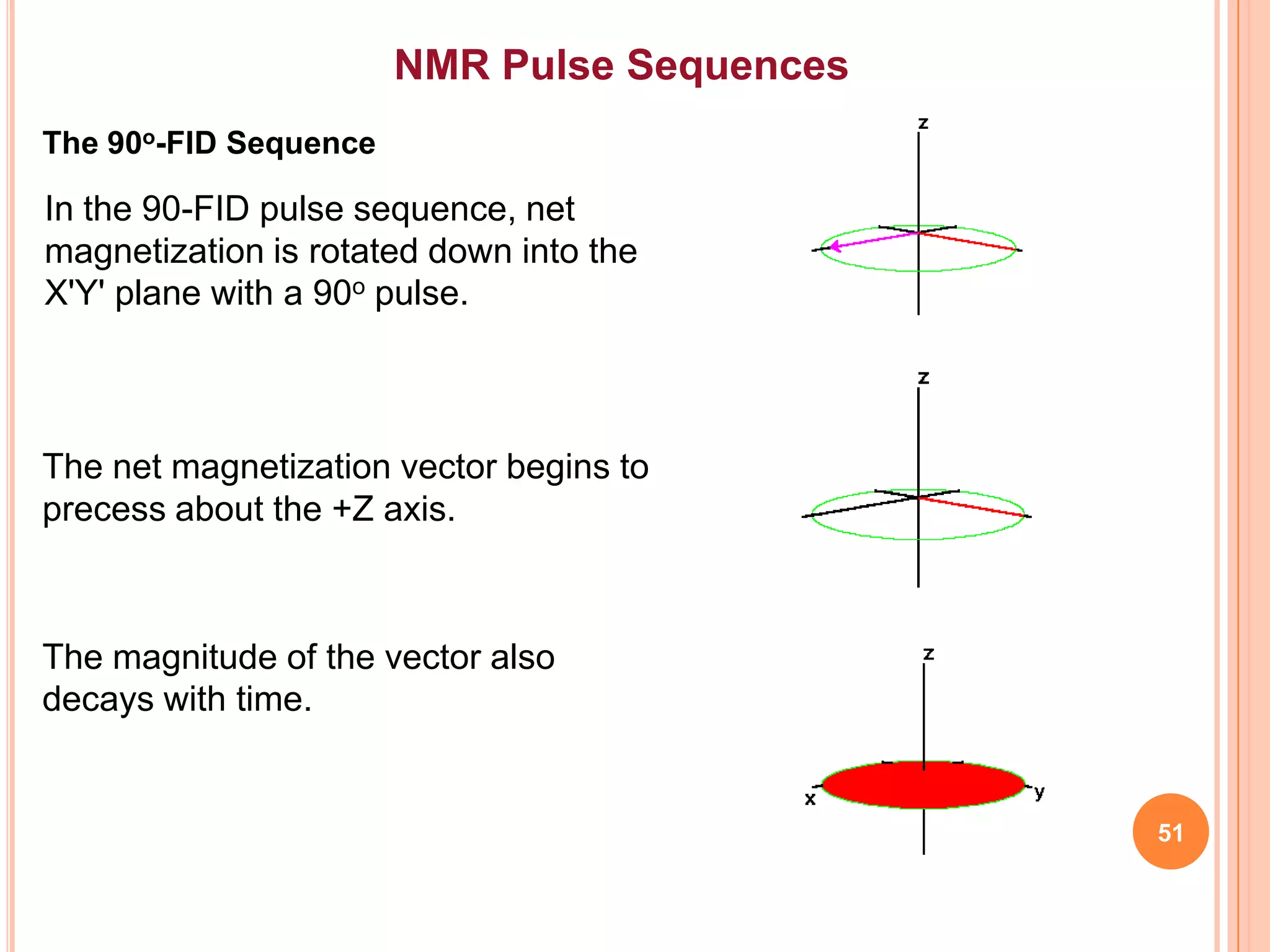
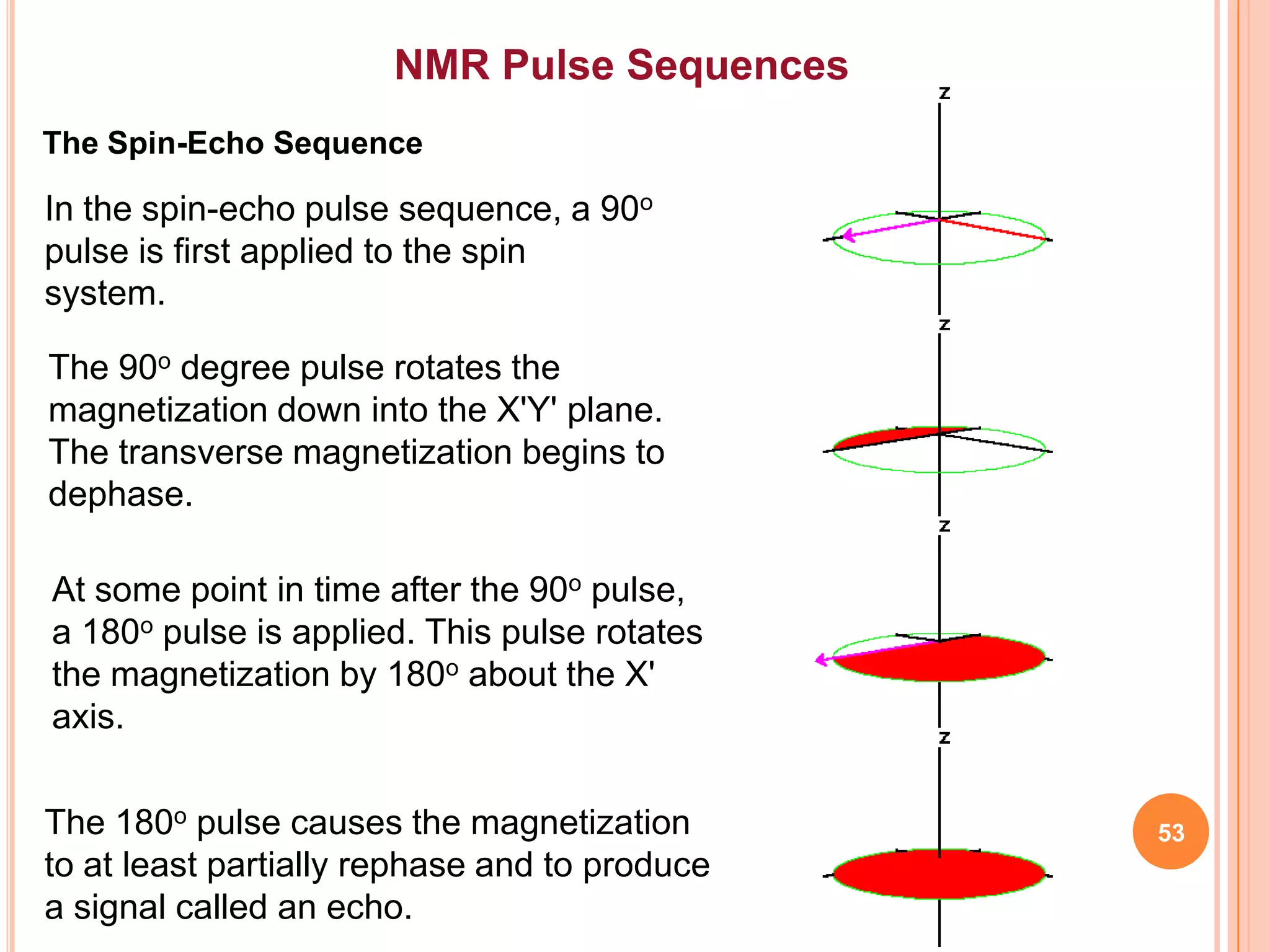
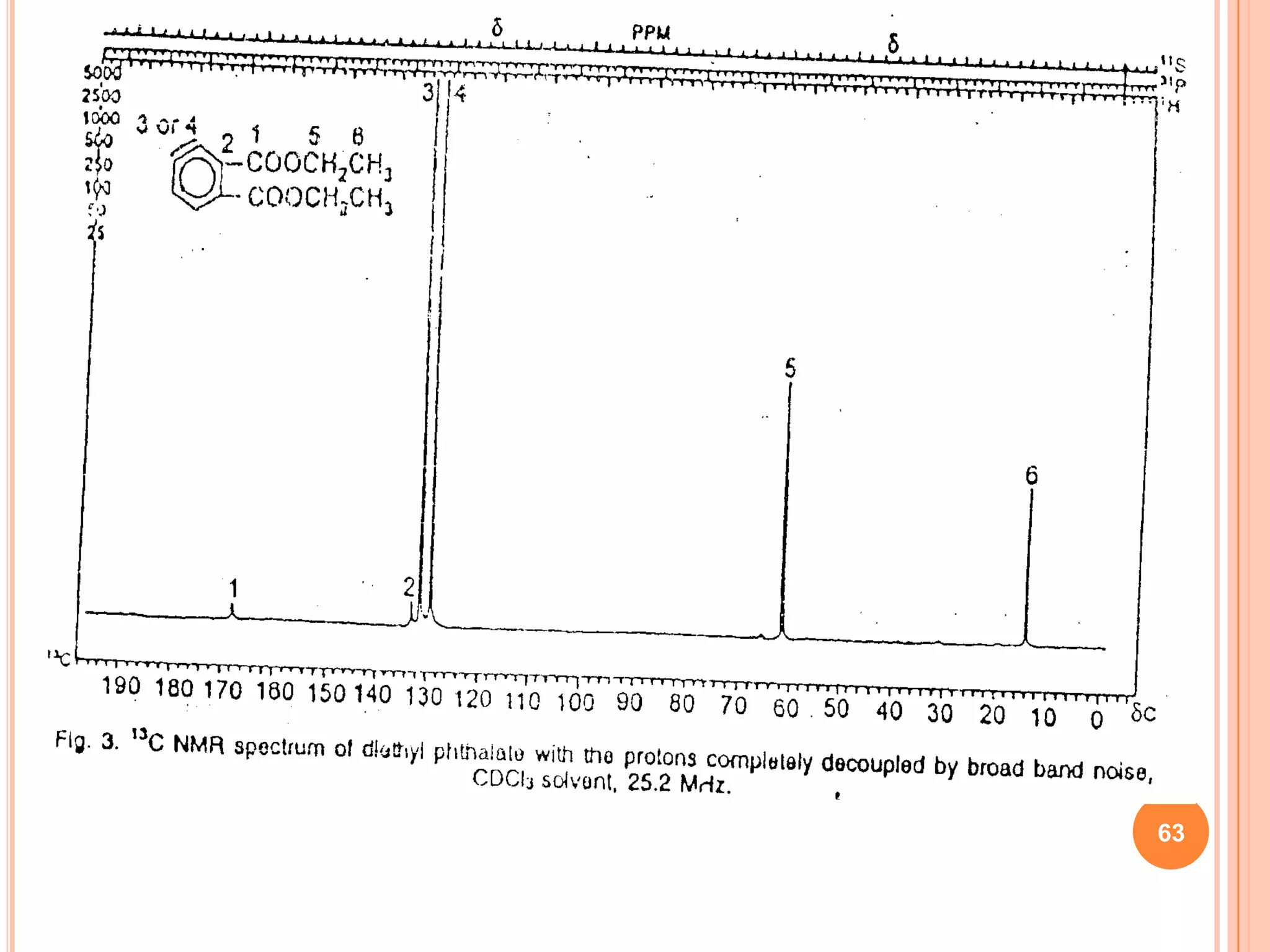
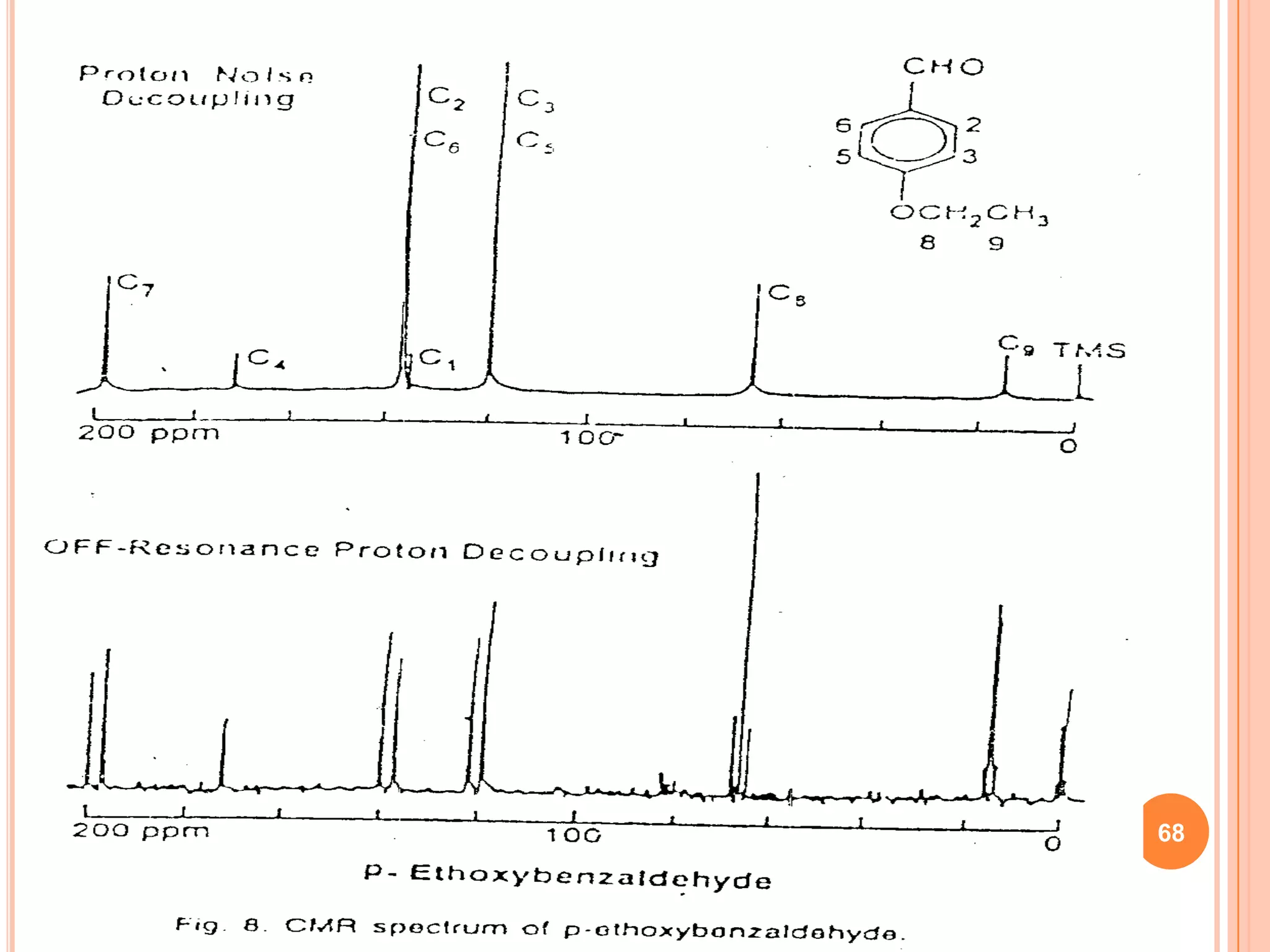
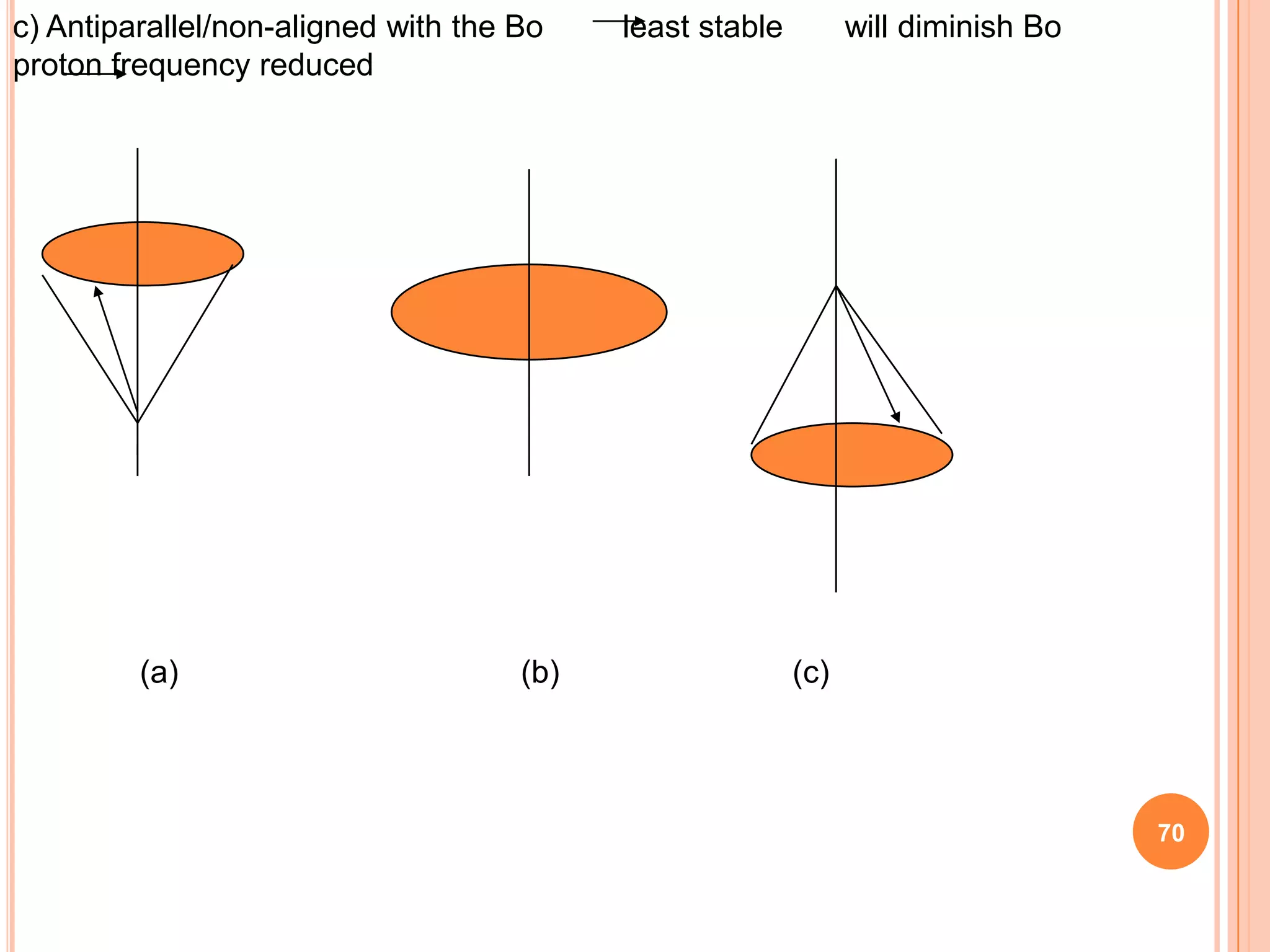
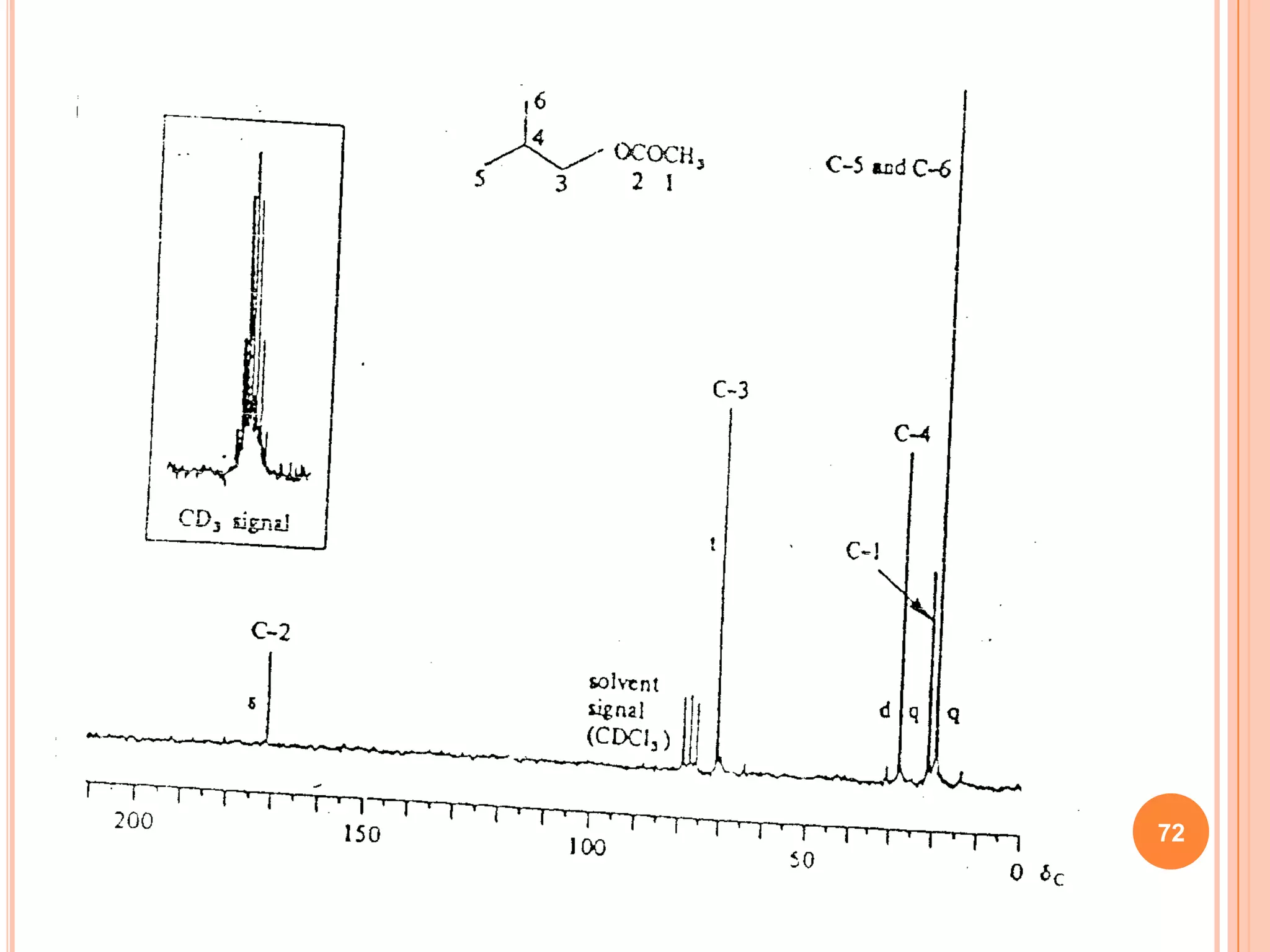
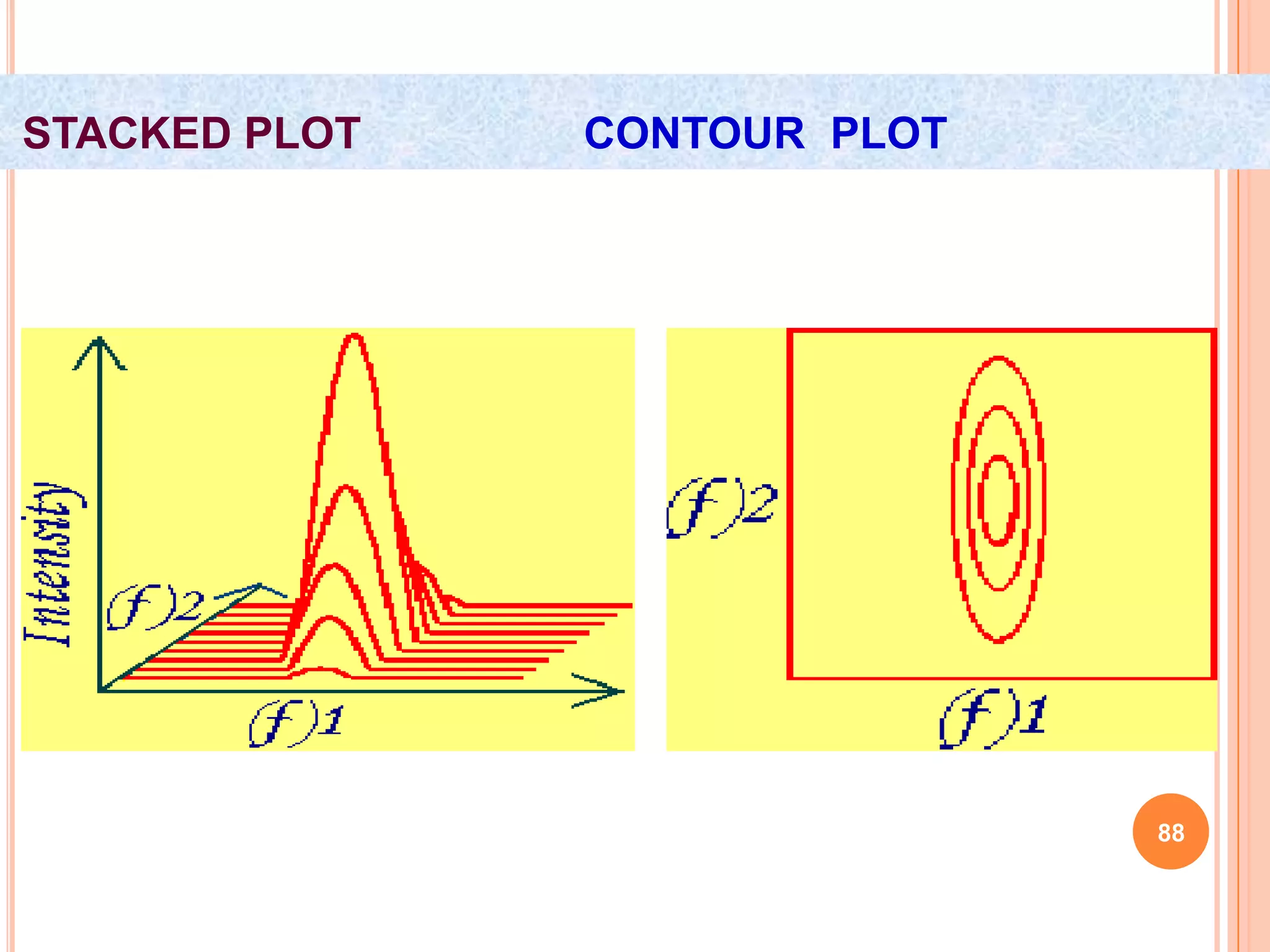
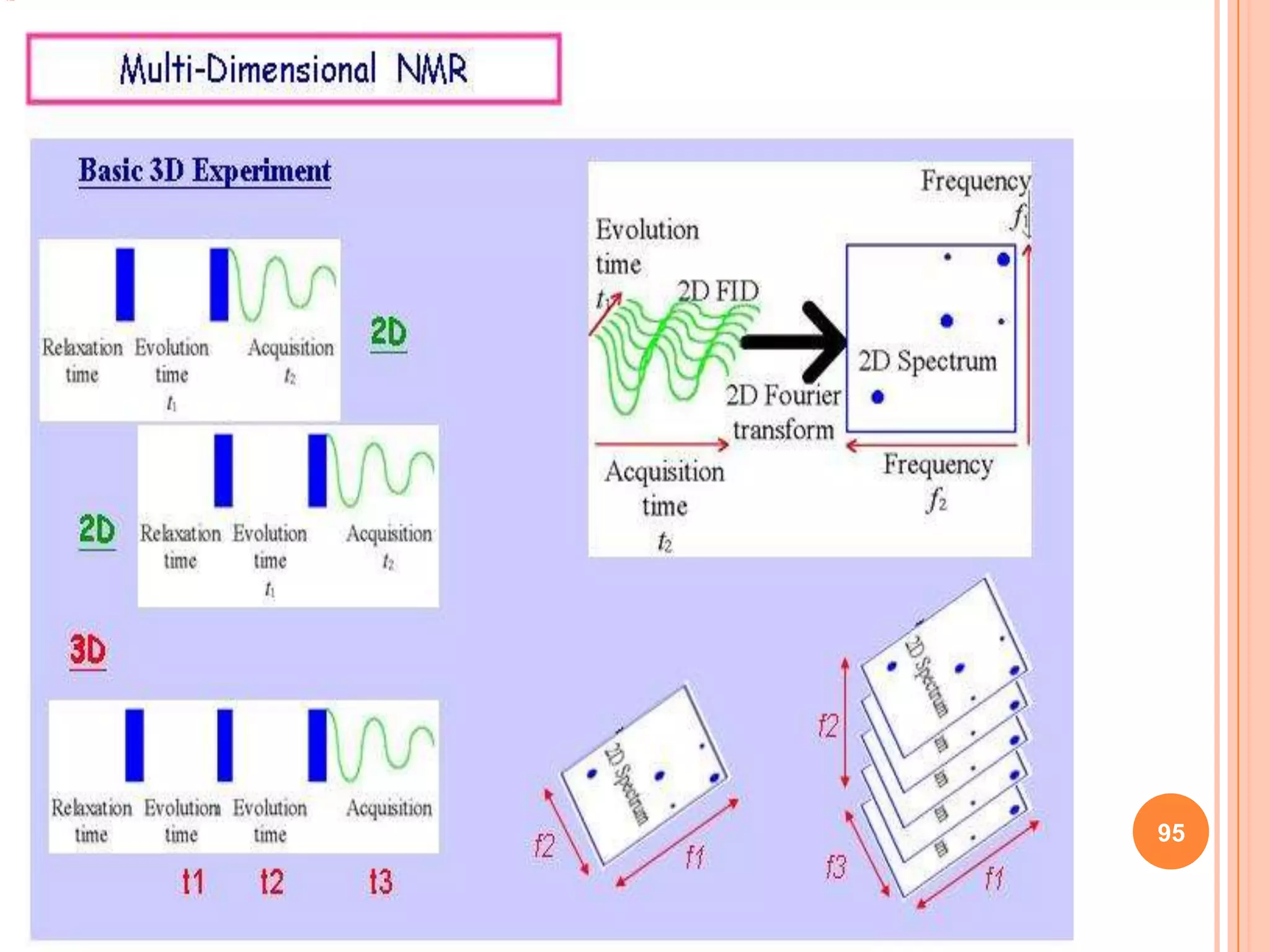
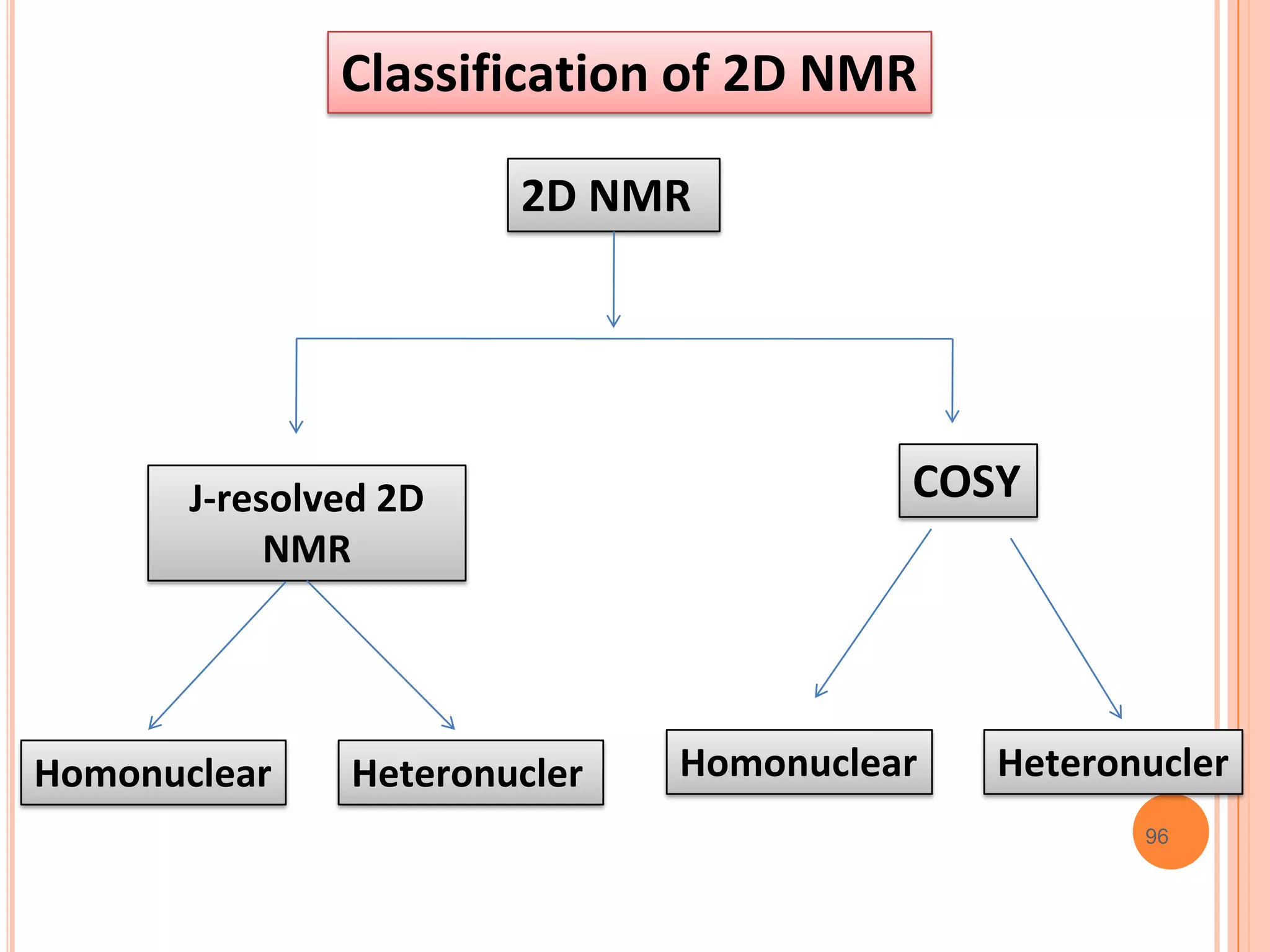
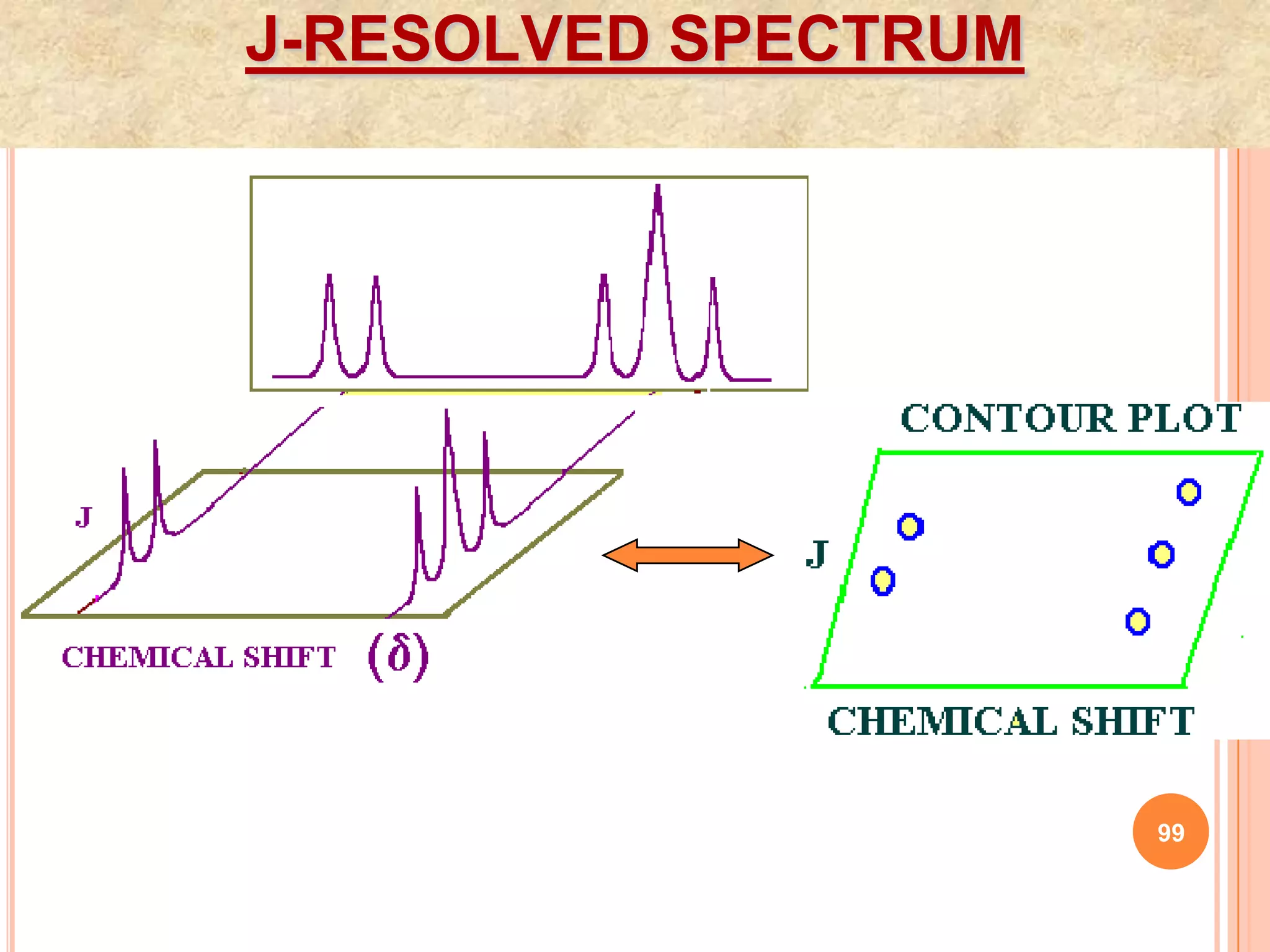
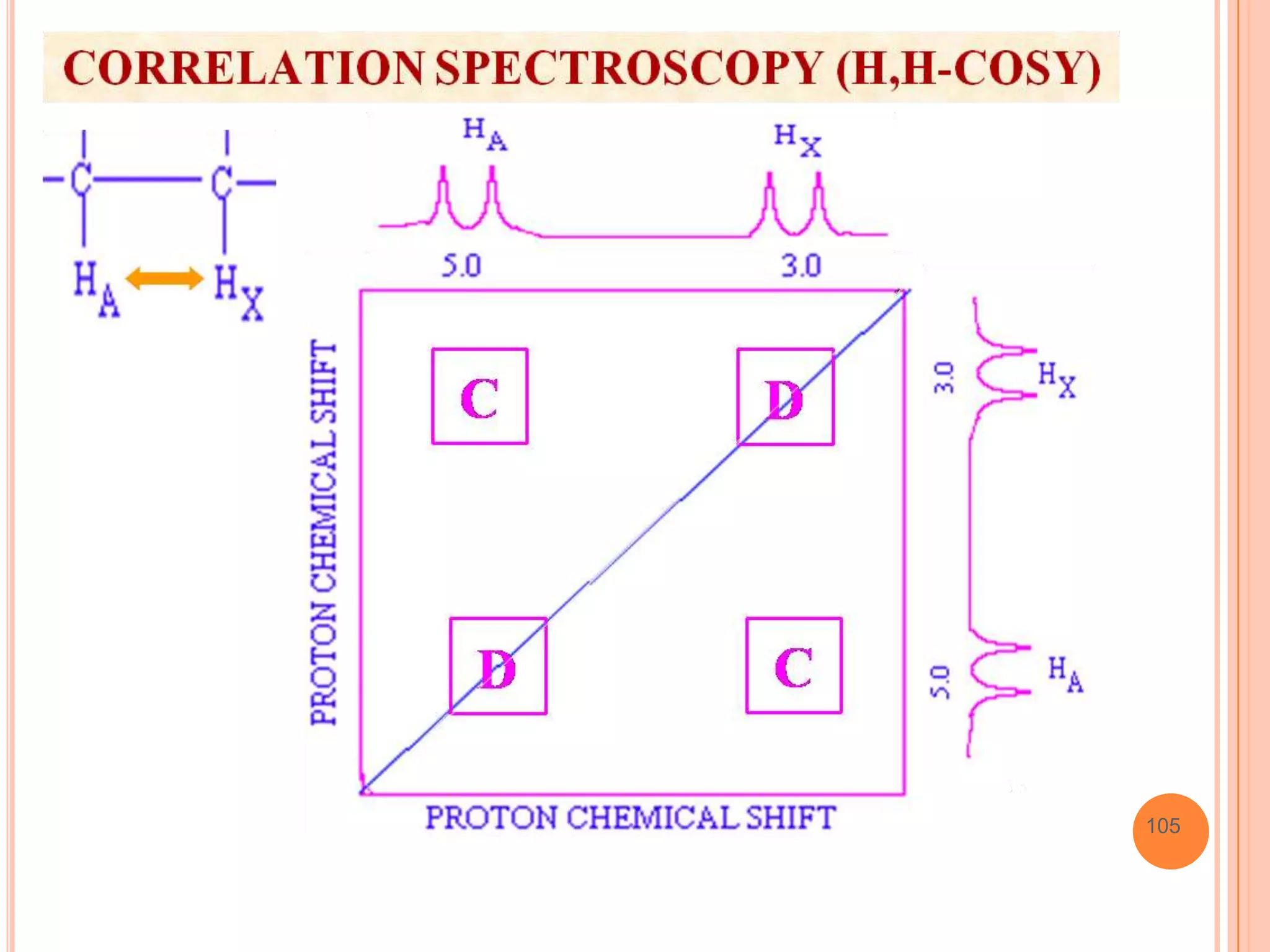
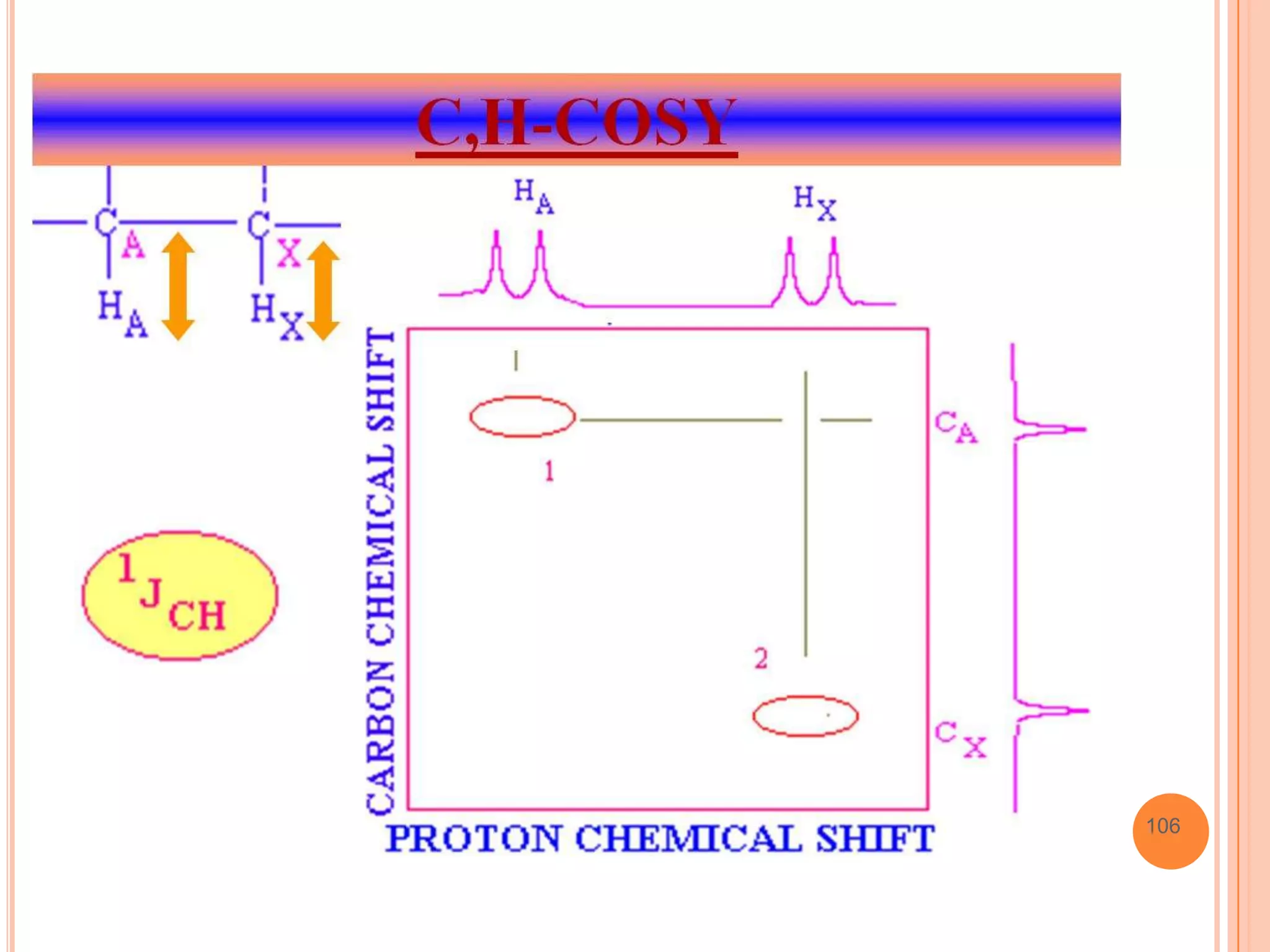
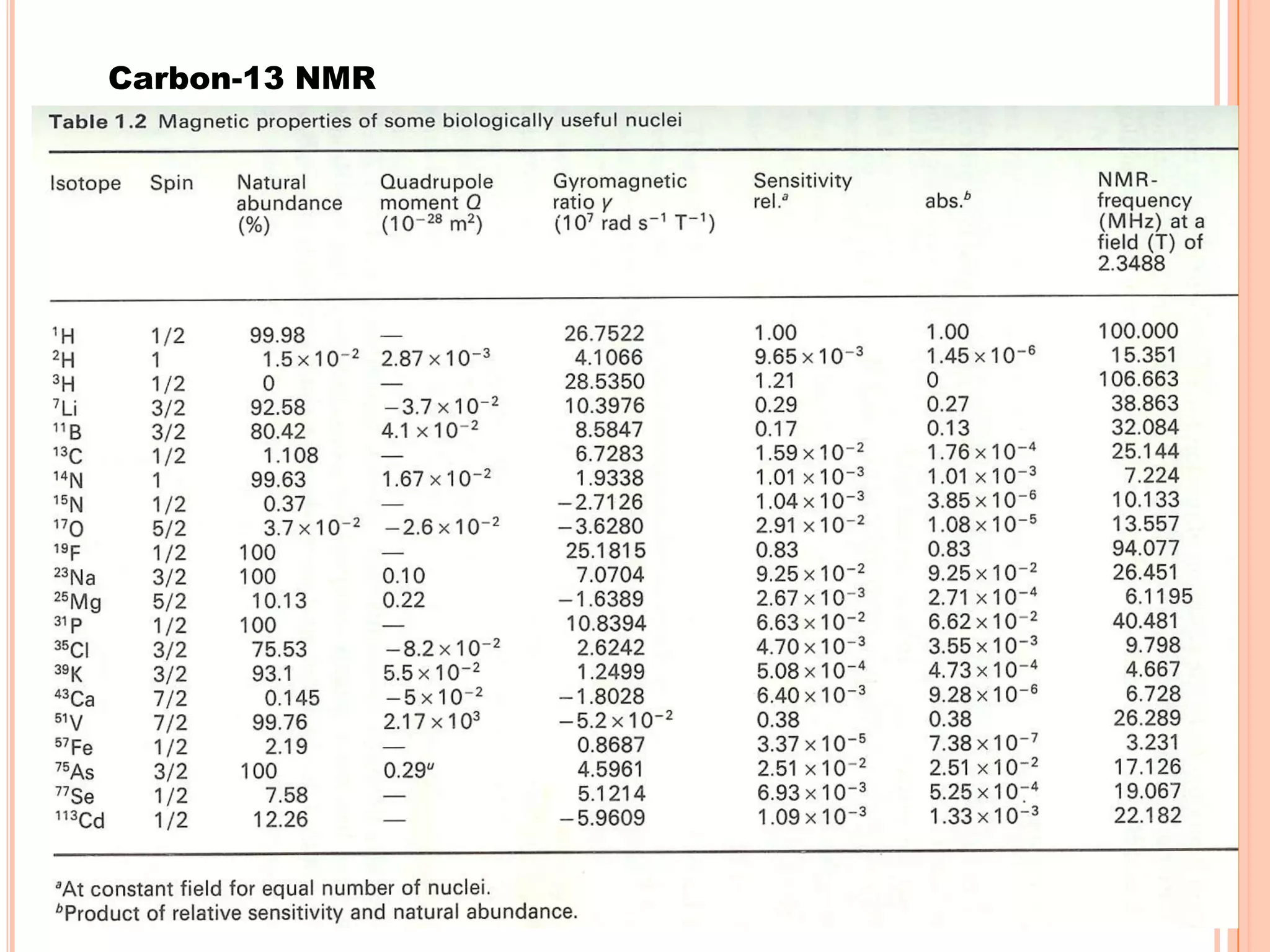
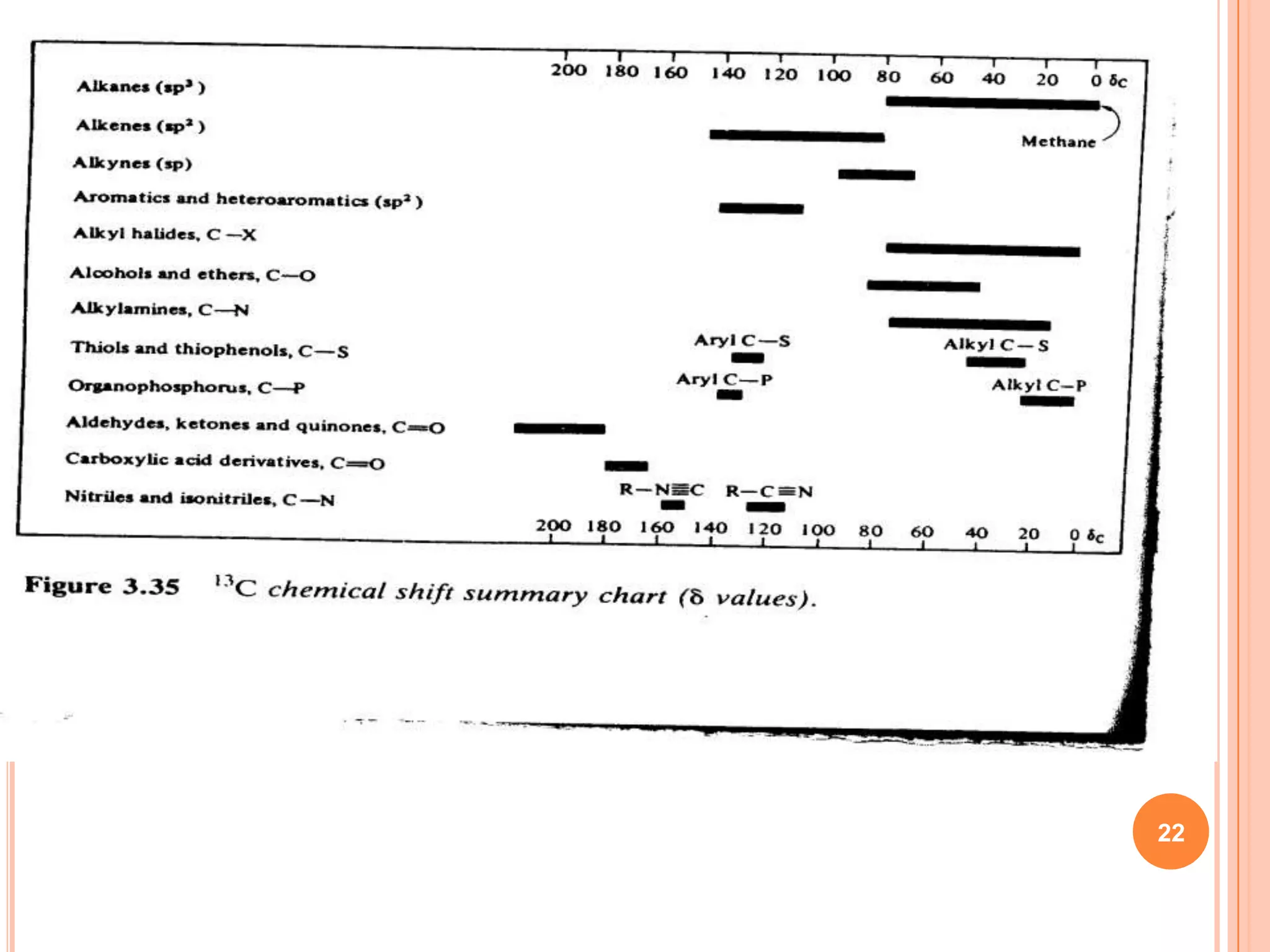
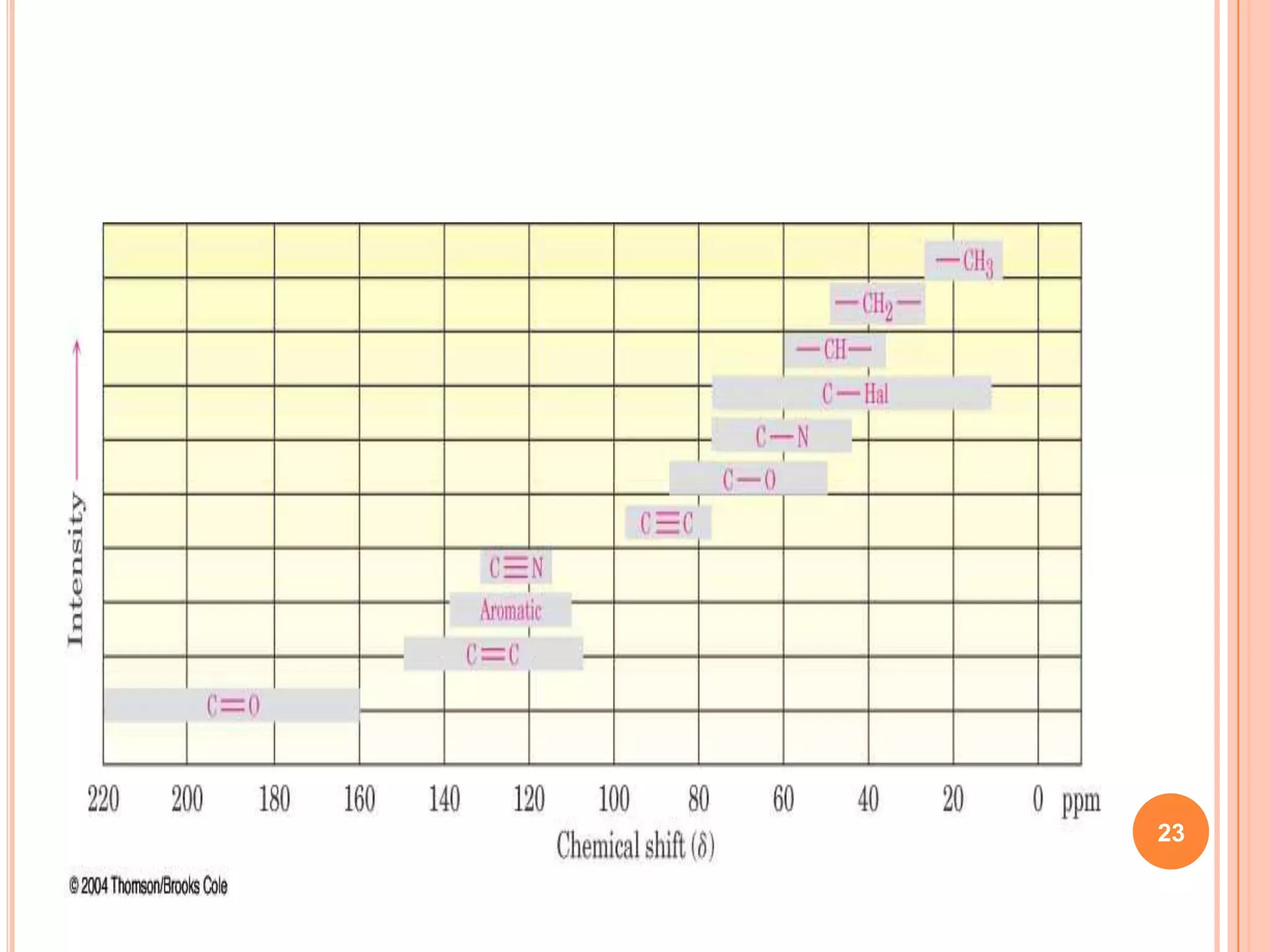
This document discusses 13C NMR spectroscopy. It begins by introducing 13C as a stable carbon isotope that can be used for NMR similarly to 1H NMR. It then covers key topics like the low natural abundance of 13C, difficulties in recording 13C spectra compared to 1H, and techniques used to overcome low sensitivity like Fourier transform NMR and decoupling. The document provides an overview of 13C NMR spectroscopy and how it can provide complementary structural information to 1H NMR.




















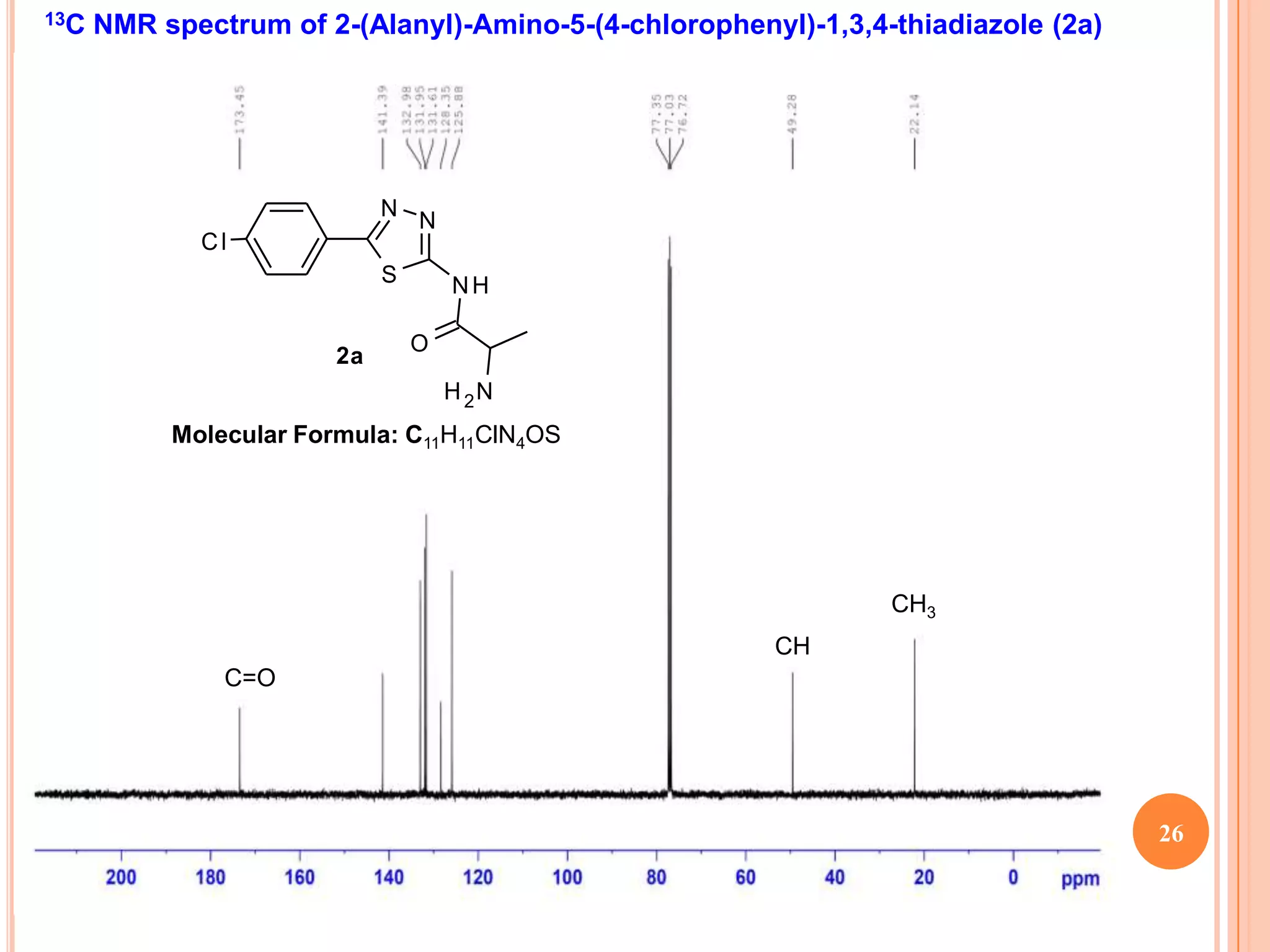
![13C NMR spectrum of 2-Amino-5-(4-methylphenyl)-5H-thiazolo[4,3-b]-1,3,4-
thiadiazole (1b)
H 3C
S
N
N S
NH2
Molecular Formula: C11H11N3S2
CH3
24](https://image.slidesharecdn.com/nmrpradip-121231004624-phpapp01/75/Nmr-pradip-24-2048.jpg)
![13C NMR spectrum of 2-(Alanyl)-Amino-5-(4-methylphenyl)-5H-thiazolo[4,3-b]-1,3,4-
thiadiazole (3c)
S
H 3C N S
N
NH
3c O
CH3
H 2N
Molecular Formula: C14H16N4OS2
CH3
C=O CH
25](https://image.slidesharecdn.com/nmrpradip-121231004624-phpapp01/75/Nmr-pradip-25-2048.jpg)
![13C NMR spectrum of 5-(4-methylphenyl)-N-[(1E)-phenylmethylene][1,3]thiazolo[4,3-
b][1,3,4]thiadiazol-2-amine (3d)
S
H 3C N S
N
3d N
M olecular F orm ula = C1 8H 1 5N 3S 2
CH3
CH
27](https://image.slidesharecdn.com/nmrpradip-121231004624-phpapp01/75/Nmr-pradip-27-2048.jpg)
![13C NMR spectrum of 3-chloro-1-[5-(4-methylphenyl)[1,3]thiazolo[4,3-b][1,3,4]thiadiazol-2-yl]-4-
phenylazetidin-2-one (4d)
S
S
N O
N N
Cl
H 3C
4d
CH3
M olecular F orm ula = C 20 H 16 C lN 3 O S 2
CH-Cl
C=O
CH
28](https://image.slidesharecdn.com/nmrpradip-121231004624-phpapp01/75/Nmr-pradip-28-2048.jpg)