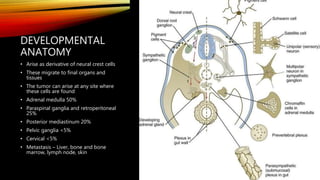
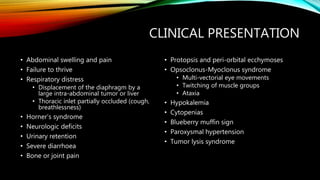
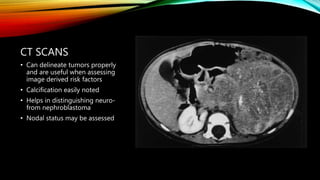
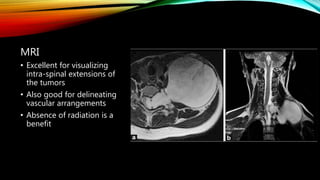
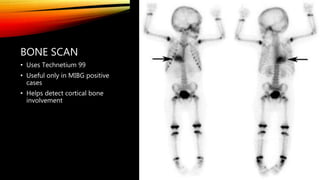
Neuroblastoma is a cancer that arises from nerve tissue and is the most common cancer in infants. It develops from immature nerve cells called neuroblasts. The majority of cases are diagnosed in children under 5 years old. Treatment is multifaceted and depends on the stage and risk classification, ranging from observation for low risk localized disease to intensive chemotherapy, surgery, radiation and stem cell transplant for high risk metastatic cancer. Despite advances, outcomes remain poor for aggressive high risk disease.











![DIAGNOSIS
• Laboratory evaluation
• Full blood count
• Renal function test
• Liver function test
• Urinalysis
• Urine assay for catecholamines [Homovanillic acid (HVA) and Vanillylmandellic acid
(VMA)]
• Tumor markers
• Neuron specific enolase
• Chromogranin A
• Ferritin](https://image.slidesharecdn.com/neuroblastoma-220815034507-8576333a/85/NEUROBLASTOMA-pptx-13-320.jpg)




























































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)