Downloaded 19 times

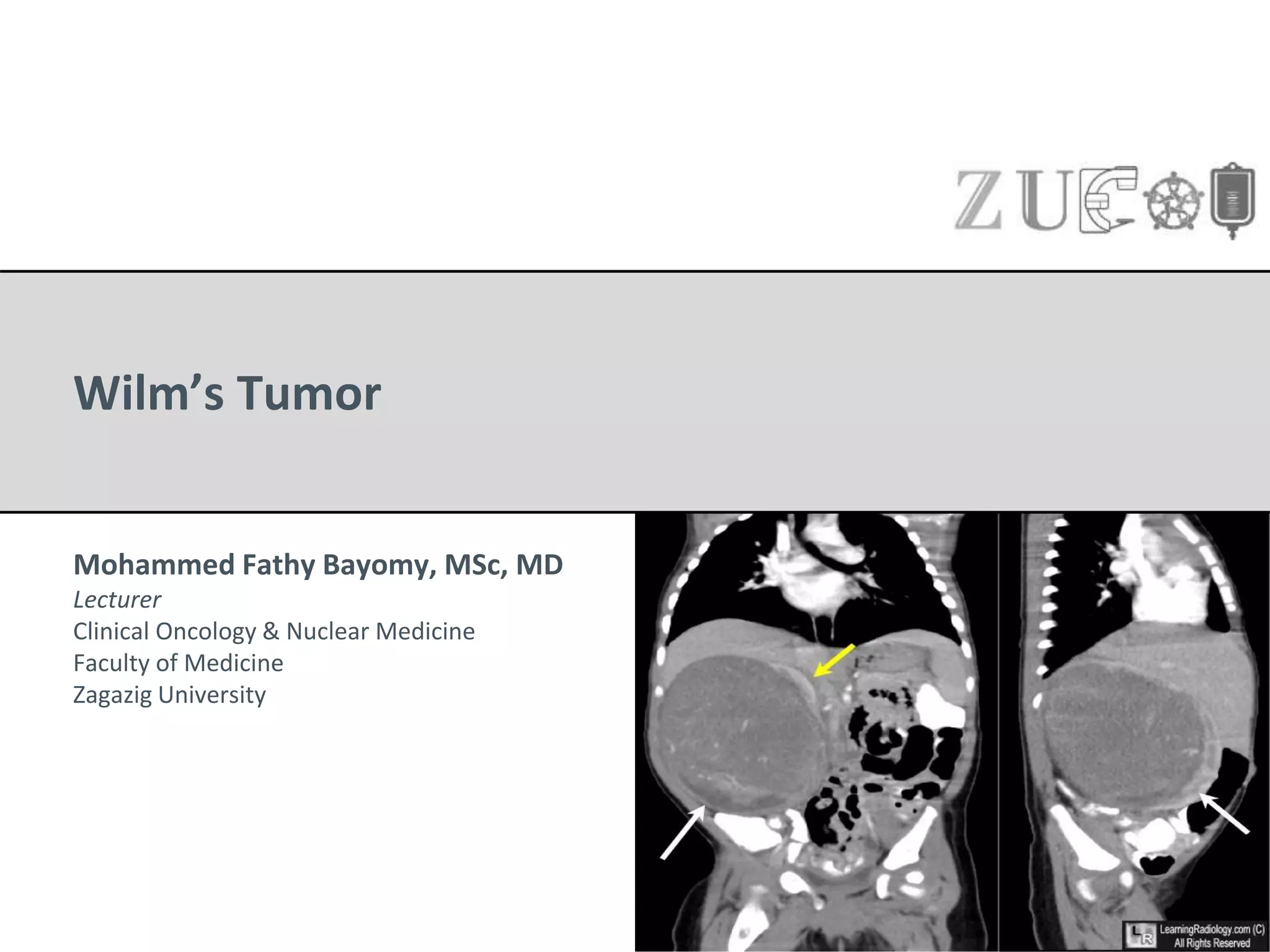















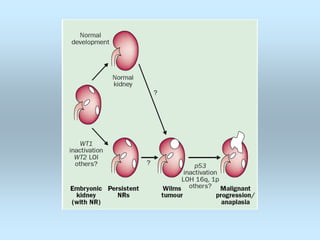

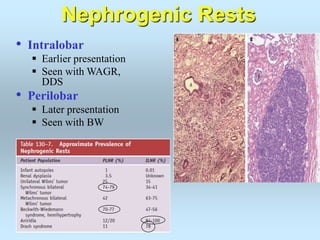



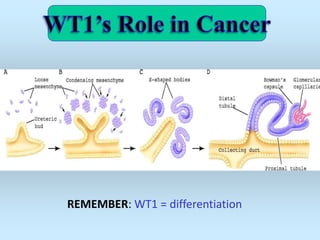







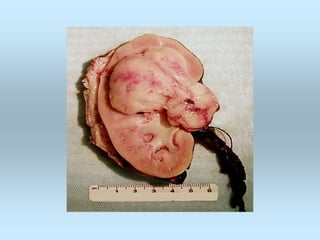
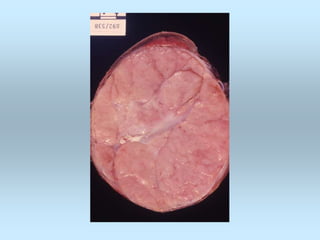
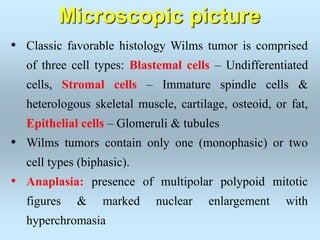
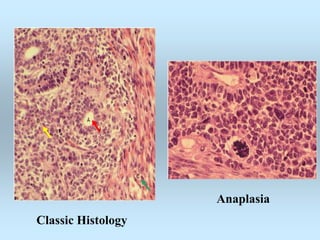


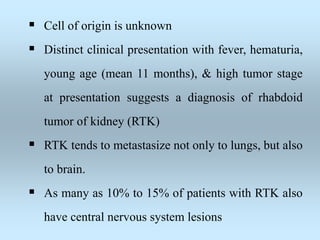


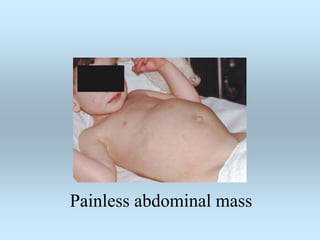




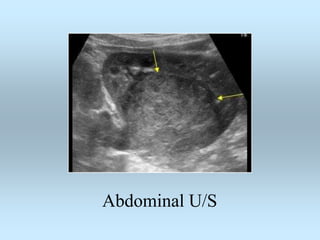

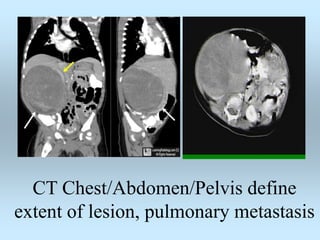
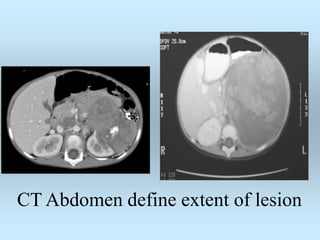
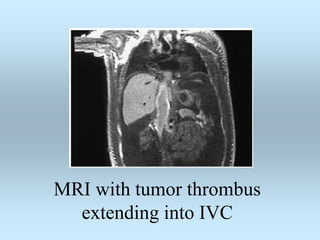

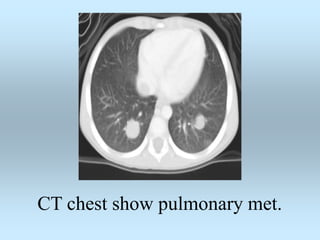
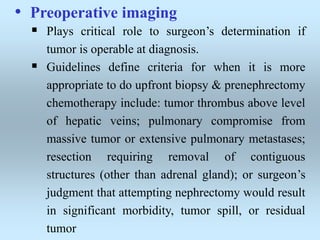



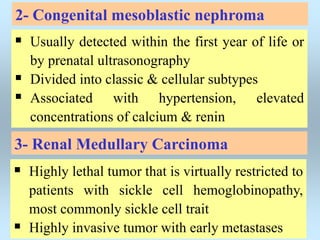








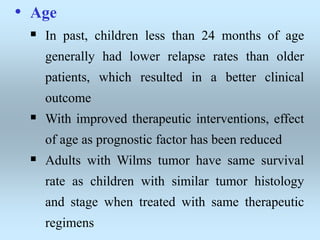
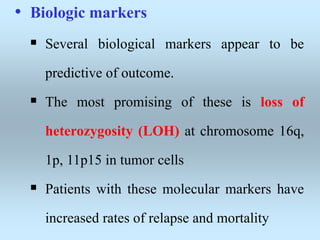

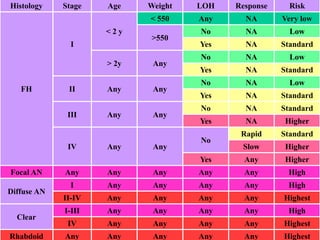



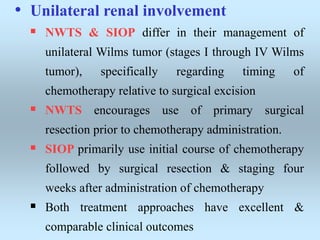



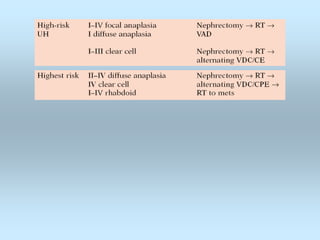
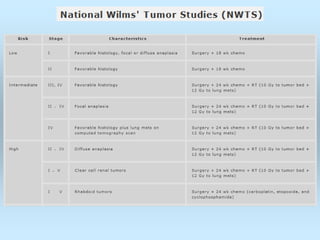













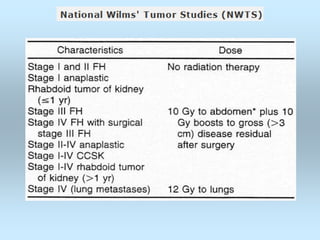
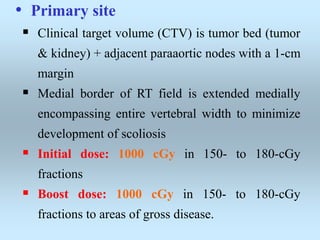
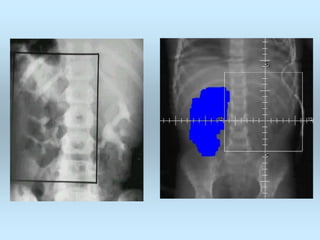

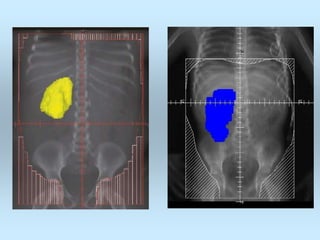

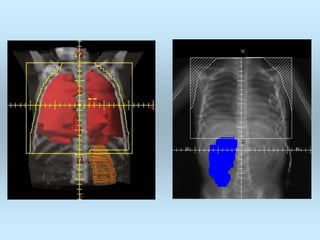

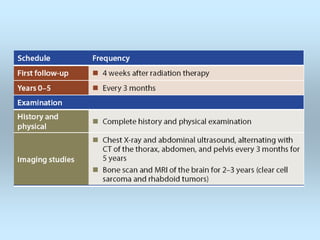

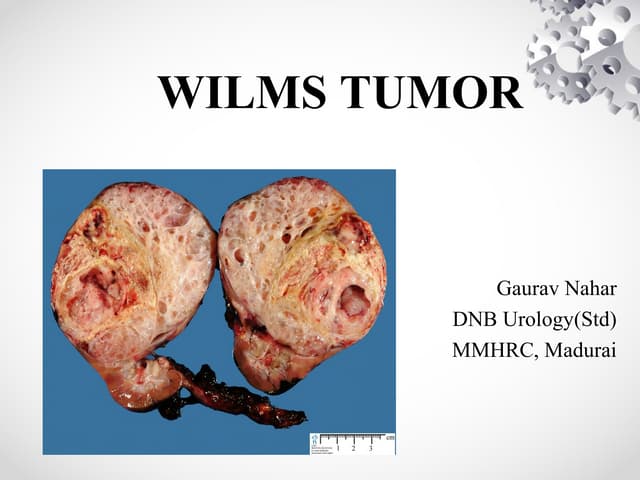
Wilm's tumor is the most common renal malignancy in children. It typically presents as a painless abdominal mass in children around 3 years old. Imaging such as ultrasound and CT are used to evaluate the tumor extent and presence of metastases. The tumor is staged surgically, with higher stages indicating a worse prognosis. Histology is also important, as the presence of anaplasia predicts a poorer outcome. Genetic conditions like WAGR and Denys-Drash syndromes increase the risk of developing Wilm's tumor. Treatment involves surgery, chemotherapy, and sometimes radiation therapy.












































![Wilm's tumour - The most common kidney tumor in children - Dr Vishnu A [VCR],...](https://cdn.slidesharecdn.com/ss_thumbnails/vishnu-wilmstumour-210312145616-thumbnail.jpg?width=640&height=640&fit=bounds)























![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)


![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)












![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)

