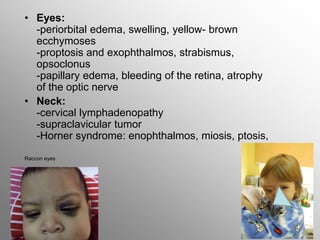
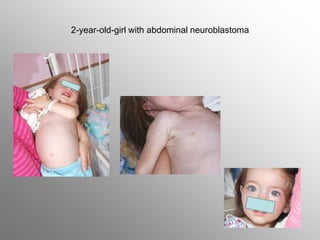
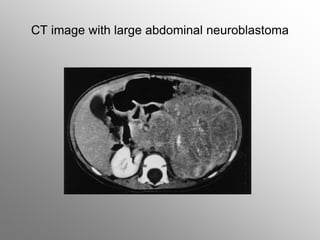
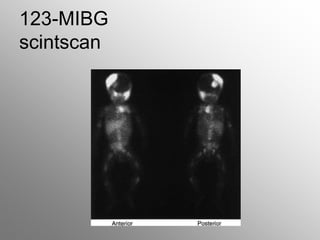
Central nervous system malignancies are the most common malignant solid tumors in childhood, accounting for 20% of cancers. The most common types are medulloblastoma, astrocytoma, ependymoma, and germinoma. Risk factors include genetic syndromes like neurofibromatosis. Symptoms depend on tumor location and type, and can include increased intracranial pressure, neurological deficits, and endocrine abnormalities. Diagnosis involves imaging like MRI and biopsy. Treatment involves surgery, radiation, and chemotherapy. Prognosis depends on tumor type and extent.
































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)






