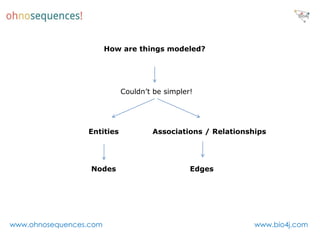
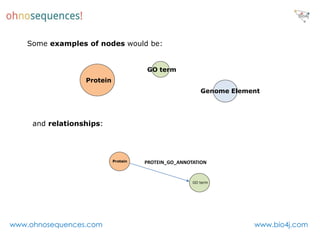
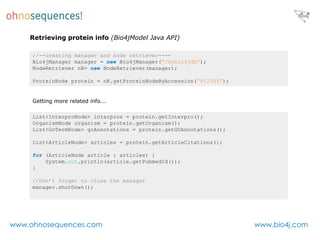
Download as PDF, PPTX














![Querying Bio4j with Cypher
Getting a keyword by its ID
START k=node:keyword_id_index(keyword_id_index = "KW-0181")
return k.name, k.id
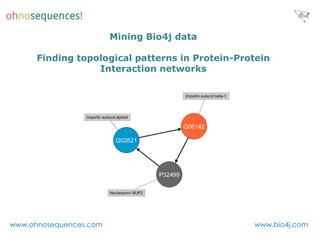
Finding circuits/simple cycles of length 3 where at least one protein is from Swiss-Prot
dataset:
START d=node:dataset_name_index(dataset_name_index = "Swiss-Prot")
MATCH d <-[r:PROTEIN_DATASET]- p,
circuit = (p) -[:PROTEIN_PROTEIN_INTERACTION]-> (p2) -
[:PROTEIN_PROTEIN_INTERACTION]-> (p3) -[:PROTEIN_PROTEIN_INTERACTION]->
(p)
return p.accession, p2.accession, p3.accession
Check this blog post for more info and our Bio4j Cypher cheetsheet
www.ohnosequences.com www.bio4j.com](https://image.slidesharecdn.com/neo4jandbioinformatics-120809173457-phpapp01/85/Neo4j-and-bioinformatics-22-320.jpg)
![A graph traversal language
Get protein by its accession number and return its full name
gremlin> g.idx('protein_accession_index')[['protein_accession_index':'P12345']].full_name
==> Aspartate aminotransferase, mitochondrial
Get proteins (accessions) associated to an interpro motif (limited to 4 results)
gremlin>
g.idx('interpro_id_index')[['interpro_id_index':'IPR023306']].inE('PROTEIN_INTERPRO').outV.
accession[0..3]
==> E2GK26
==> G3PMS4
==> G3Q865
==> G3PIL8
Check our Bio4j Gremlin cheetsheet
www.ohnosequences.com www.bio4j.com](https://image.slidesharecdn.com/neo4jandbioinformatics-120809173457-phpapp01/85/Neo4j-and-bioinformatics-23-320.jpg)











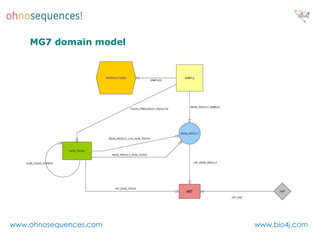

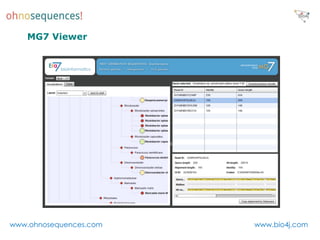
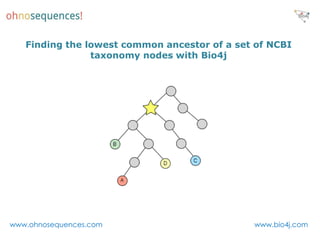




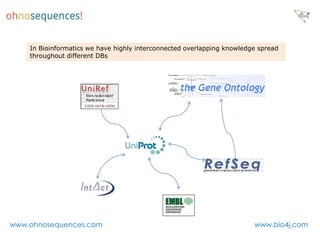
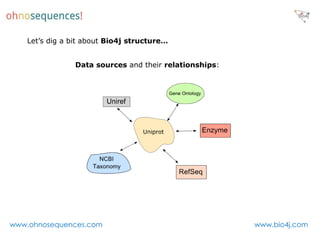
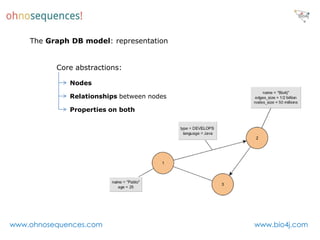
This document discusses Neo4j and its applications in bioinformatics. It describes Bio4j, an open source bioinformatics graph database built using Neo4j that integrates data from sources like Uniprot, NCBI taxonomy, Gene Ontology, and more. Bio4j models biological data as nodes and relationships in a graph structure rather than tables. This allows for more flexible querying and knowledge integration. The document provides examples of how Bio4j can be accessed through its Java API, Cypher query language, Gremlin traversal language, and REST API. It also describes some tools and visualizations for exploring and analyzing Bio4j data.






































































![5G Explained! A High Level Overview [Introduction]](https://cdn.slidesharecdn.com/ss_thumbnails/5gexplainedahighleveloverview-260119165306-cc137a3e-thumbnail.jpg?width=640&height=640&fit=bounds)
















