Mucopolysachridosis
•Download as PPTX, PDF•
43 likes•11,587 views

Mucopolysaccharidoses are hereditary lysosomal storage disorders caused by deficiencies in enzymes that break down glycosaminoglycans. This results in glycosaminoglycan accumulation in tissues and organs, leading to symptoms like facial abnormalities, short stature, corneal clouding, developmental delays, bone deformities, organ enlargement, and neurological issues. Diagnosis involves testing urine for glycosaminoglycan levels and measuring enzyme activity levels. Treatment focuses on managing symptoms through supportive care, physical therapy, surgeries, and enzyme replacement therapy or hematopoietic stem cell transplantation to reduce clinical effects.

More Related Content
What's hot
What's hot (20)
Similar to Mucopolysachridosis
Similar to Mucopolysachridosis (20)
Recently uploaded
Recently uploaded (20)
Mucopolysachridosis
- 2. Contents Introduction Clinical presentation Types Management
- 3. Introduction
- 4. Introduction Mucopolysaccharidoses are hereditary, progressive diseases caused by mutations of genes coding for lysosomal enzymes leading to defects in stepwise breakdown of glycosaminoglycans (GAGs). Glycosaminoglycans _formally named mucopolysaccharides_ are large, complex polymers of linear repeating sulfated acidic and amino sugar disaccharide units widely distributed in most of the tissues. Neufeld E, Muenzer J. The mucopolysaccharidoses.The Metabolic and Molecular Basis of Inherited Disease. 8 ed. New York. NY: McGraw-Hill; 2001:3421-52
- 5. Introduction For examples; they are components of the ground substance of bone and cartilage, lubricant in joint fluid, and the surface coating that initially binds growth factors to cells. The metabolic recycling of GAGs requires the stepwise degradation of the terminal sulfate, acidic, and amino sugar residues by a series of lysosomal enzymes. The deficiency of one of these enzymes blocks degradation of the substrate and results in a specific disorder.
- 6. Mode of Inheritance All types of MPS are autosomal recessive disorders except type II (Hunter syndrome) which is X-linked recessive. The estimated total incidence of all types of MPS of approximately 1 in 20,000 live births. The most common subtype is MPS-III followed by MPS-I and MPS-II Neufeld E, Muenzer J. The mucopolysaccharidoses.The Metabolic and Molecular Basis of Inherited Disease. 8 ed. New York. NY: McGraw-Hill; 2001:3421-52
- 8. Classification contd… According to their dominant clinical features MPSs can be grouped into four broad categories: Soft tissue storage and skeletal disease with or without brain disease (MPS I, II, VII). Soft tissue and skeletal disease (MPS VI) Primarily skeletal disorders (MPS IVA, IVB) Primarily central nervous system disorders (MPS III A-D) Neufeld E, Muenzer J. The mucopolysaccharidoses.The Metabolic and Molecular Basis of Inherited Disease. 8 ed. New York. NY: McGraw-Hill;
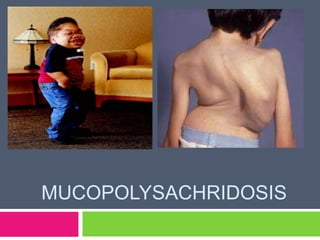
- 10. Clinical Presentation The mucopolysaccharidoses share many clinical features but have varying degrees of severity depending on the mucopolysaccharidosis subtype. These features may not be apparent at birth but progress as storage of glycosaminoglycans increases with time affecting bone, skeletal structure, connective tissues, and brain and internal organs.
- 11. Common Presentations Mental retardation Developmental delay Severe behavioral problems Corneal clouding Coarse facial features Short stature ,short trunk Skeletal irregularities Neufeld E, Muenzer J. The mucopolysaccharidoses.The Metabolic and Molecular Basis of Inherited Disease. 8 ed. New York. NY: McGraw-Hill; 2001:3421-52
- 12. Contd.. Hepatosplenomegaly Hernias Joint stiffness Hydrocephalus Dysostosis multiplex Cardiac disease Retinal degeneration Hair loss or Hirsutism
- 15. Cardiac manifestation IH (Hurler) Most severe - heart muscle thick/weak Heart vessels thickened - heart attack/angina Thickened/nodular leaky valves - mitral valve/aortic valve IS (Scheie) Involve heart valves - aortic/mitral II (Hunter) Heart Vessels - Heart attack Thickened valves - mitral/aortic leak Gatzoulis M A , et al: Cardiac involvement in mucopolysaccharidoses: effects of allogeneic bone marrow transplantation. Arch Dis Child. 1995 September; 73(3): 259–260.
- 16. IIIA (Sanfilippo) Aortic valve leak, mitral valve leak IVA (Morquio) Heart wall, thickened/leaky valves - aortic/mitral VI (Maroteaux-Lamy) Heart wall thickened/stiff, weak Thickened/narrow/leaky valves - mitral/aortic valves VII (Sly) Leaky valve aortic Gatzoulis M A , et al: Cardiac involvement in mucopolysaccharidoses: effects of allogeneic bone marrow transplantation. Arch Dis Child. 1995 September; 73(3): 259–260.
- 17. Clinical presentation Coarse facial features (flat nasal bridge, thick lips and large tongue, Prominent forehead) Moderate corneal opacifation Gatzoulis M A , et al: Cardiac involvement in mucopolysaccharidoses: effects of allogeneic bone marrow transplantation. Arch Dis Child. 1995 September; 73(3): 259–260.
- 18. Clinical Presentation Sever kyphoscliosis in patient with Morquio disease (MPS IV) Gatzoulis M A , et al: Cardiac involvement in mucopolysaccharidoses: effects of allogeneic bone marrow transplantation. Arch Dis Child. 1995
- 19. Dysostosis multiplex In patient with MPS type VI: A, B) hands of patients at the age of 7 and 16 years : deformity and shortening of metacarpal bones. C, D) the spine of patient at the age of 11 and 16 years : scoliosis, abnormal shape of the vertebral bodies. E, F) the pelvis of patients at the age of 11 and 16 years : irregular shape of the pelvis, hypoplastic hip acetabulum, lopsided head of hip bones.Gatzoulis M A , et al: Cardiac involvement in mucopolysaccharidoses: effects of allogeneic bone marrow transplantation. Arch Dis Child. 1995
- 20. Types
- 21. MPS I (Hurler) Deficiency of α-L-iduronidase Divided into three subtypes: 1) Hurler disease: Is a severe, progressive disorder with multiple organ and tissue involvement that results in premature death, usually by 10 years of age. Clinical features: •Corneal clouding •Dysostosis multiplex •Hepatosplenomegal y • Mental retardation •Cardiomyopathy • Coarse facial features Clinical features and diagnosis of the mucopolysaccharidoses, Uptodate, http://www.uptodate.com/contents/clinical-features-and-diagnosis-of-the-mucopolysaccharidoses
- 22. MPS I (Hurler) 2) Hurler-Scheie Disease Progresses less rapidly than Hurler syndrome. The onset of symptoms is usually observed between 3 and 8 yr of age. Patients typically die in their twenties of cardiac disease or respiratory failure Clinical features: Joint stiffness is the most presenting features Usually have normal intelligence Cardiac disease Dysostosis multiplex. Clinical features and diagnosis of the mucopolysaccharidoses, Uptodate, http://www.uptodate.com/contents/clinical-features-and-diagnosis-of-the-mucopolysaccharidoses
- 23. 3) Scheie syndrome least severe form of MPS I, Most die in their middle decades with cardiac disease Late diagnosis (teenage years) Clinical features: Most presenting features are joint stiffness and corneal clouding Hydrocephalus, optical nerve compression Aortic valve disease MPS I (Hurler) Clinical features and diagnosis of the mucopolysaccharidoses, Uptodate, http://www.uptodate.com/contents/clinical-features-and-diagnosis-of-the-mucopolysaccharidoses
- 24. MPS II (Hunter) Deficiency of iduronate-2-sulfatase. Is an X-linked disorder Manifests almost exclusively in males (except in case of skewed inactivation of the X chromosome carrying the normal gene). Clinical features and diagnosis of the mucopolysaccharidoses, Uptodate, http://www.uptodate.com/contents/clinical-features-and-diagnosis-of-the-mucopolysaccharidoses
- 25. Clinical features: The severe form shares features with Hurler syndrome except of slow progression and absence of corneal clouding. Patients with the mild form have minimal CNS involvement, slow progression of somatic deterioration and preservation of intelligence in adult life. In mild form, survival to ages 65 and 87 yr has been reported in mild form.
- 26. MPS III (Sanfilippo syndrome) Most common type of all MPS Divided into four subtypes (A-D).Each type is caused by a different enzyme deficiency involved in the degradation of heparan sulfate Clinical features: Progressive dementia Aggressive behavior Hyperactivity, Sleep disorders Clinical features and diagnosis of the mucopolysaccharidoses, Uptodate, http://www.uptodate.com/contents/clinical-features-and-diagnosis-of-the-mucopolysaccharidoses
- 27. Communicating hydrocephalus Spasticity , Seizures Deafness and loss of vision Mild somatic involvement ( splenomegaly, skeletal deformities)
- 28. MPS IV (Morquio disease) Subtyped into two categories: 1) MPS IV-A: N-acetylgalactosamine-6-sulfatase deficiency. 2) MPS IV-B: β-galactosidase deficiency. Both result in the defective degradation of keratan sulfate Clinical features and diagnosis of the mucopolysaccharidoses, Uptodate, http://www.uptodate.com/contents/clinical-features-and-diagnosis-of-the-mucopolysaccharidoses
- 29. Clinical features: Severe skeletal dysplasia (Dysostosis multiplex occurs early). Cervical cord compression may occur after minor falls. Short-trunk dwarfism (<125 cm). Preservation of intelligence. Mild somatic involvement.
- 30. MPS VI (Maroteaux-Lamy ) Arylsulfatase B deficiency and subsequent deposition of dermatan sulfate, and chondroitin 4-sulfate Occurs in mild, intermediate, and severe forms Clinical features: Similar to hurler in somatic manifestations. Normal intelligence. Marked corneal clouding. valvular disorders Clinical features and diagnosis of the mucopolysaccharidoses, Uptodate, http://www.uptodate.com/contents/clinical-features-and-diagnosis-of-the-mucopolysaccharidoses
- 31. MPS VII (Sly syndrome) Deficiency of β-glucuronidase Hydrops fetalis is a common presentation in sever form Clinical features: Similar to MPS I with significant soft tissue and skeletal abnormalities Dysostosis multiplex Corneal clouding
- 32. Management
- 33. Diagnosis Clinical feature: MPS disorder should be suspected in a child with coarse facial features, bone disease, developmental delay, short stature, hepatosplenomegaly, hernia, corneal clouding. Skeletal radiographs: Dysostosis multiplex GAG concentration: Measurement of urinary GAG concentration, electrophoresis. Seyrantepe V, Tihy F, Pshezhetsky AV. The microcellmediated transfer of human chromosome 8 restores the deficient Nacetylytransferase activity in skin fibroblasts of Mucopolysaccharidosis type IIIC patients. Hum Genet. Sep 2006;120(2):2936.
- 34. Diagnosis contd.. Enzyme activity assay: The definitive diagnosis of MPS requires of, usually in peripheral blood leukocytes Prenatal diagnosis: Offered for selected family Seyrantepe V, Tihy F, Pshezhetsky AV. The microcellmediated transfer of human chromosome 8 restores the deficient Nacetylytransferase activity in skin fibroblasts of Mucopolysaccharidosis type IIIC patients. Hum Genet. Sep 2006;120(2):2936.
- 35. Management Evaluations Following Initial Diagnosis: Thorough Developmental assessment. Skeletal survey: to determine the involvement of the spine and degree and extent of joint involvement. Cardiac evaluation with echocardiography.
- 36. Brain MRI, including assessment of possible hydrocephalus. Hearing assessment Ophthalmologic examination Assessment of spinal cord and peripheral nerveSeyrantepe V, Tihy F, Pshezhetsky AV. The microcellmediated transfer of human chromosome 8 restores the deficient Nacetylytransferase activity in skin fibroblasts of Mucopolysaccharidosis type IIIC patients. Hum Genet. Sep 2006;120(2):2936.
- 37. Management Treatment of Manifestations: Supportive management can improve the quality of life for affected individuals and their families. Skeletal manifestation : Physical therapy is a critical aspect of MPS therapy, range of motion exercises appear to offer some benefits in preserving joint function. Valvular disease: Cardiac valve replacement should be considered
- 38. Hydrocephaly: Ventriculoperitoneal shunting improve the quality of life . spinal cord compression: Early surgical intervention prevent severe complications Corneal clouding : Corneal transplantation is successful for individuals with attenuated disease, although donor grafts eventually become cloudy Seyrantepe V, Tihy F, Pshezhetsky AV. The microcellmediated transfer of human chromosome 8 restores the deficient Nacetylytransferase activity in skin fibroblasts of Mucopolysaccharidosis type IIIC patients. Hum Genet. Sep 2006;120(2):2936.
- 39. Enzyme-replacement therapy (ERT): Currently (ERT) available for MPS type I ,II and VI. (ERT) Has proven useful in reducing somatic symptoms and pain but show no improvement in neurological involvement as enzymes can not cross the blood brain barrier. Management
- 40. Management Hematopoietic Stem Cell Transplantation (HSCT) (HSCT) procedure carries a high risk of morbidity and mortality Pulmonary and cardiac complications post-HSCT appear to be significant Despite the high risk of procedure, HSCT has been successful in reducing the progression of some findings in children with severe MPSI Successful HSCT reduces facial coarseness, and hepatosplenomegaly, improves hearing, airway obstruction and maintains normal heart function Ross CJ, Bastedo L, Maier SA, Sands MS, Chang PL. Treatment of a lysosomal storage disease, mucopolysaccharidosis VII, with microencapsulated recombinant cells. Hum Gene Ther. Oct 10 2000;11(15):211727
- 41. Management The skeletal manifestations and corneal clouding continue to progress at the same rate in children treated with HSCT and in untreated children Children showing significant cognitive impairment prior to undergoing HSCT and ERT do not show correction of existing impairment. Survival is increased roughly 67% compared to those not receiving HSCT Ross CJ, Bastedo L, Maier SA, Sands MS, Chang PL. Treatment of a lysosomal storage disease, mucopolysaccharidosis VII, with microencapsulated recombinant cells. Hum Gene Ther. Oct 10 2000;11(15):211727
- 42. Summary Mucopolysaccharidoses (MPS) are lysosomal storage disorders caused by the deficiency of enzymes required for breakdown of glycosaminoglycans (GAGs). GAGs accumulate in the lysosomes, resulting in cellular dysfunction and clinical abnormalities. An MPS disorder should be suspected in a child with coarse facial features, short stature, corneal clouding, developmental delay, bone disease (dysostosis multiplex),
- 43. Biochemical evaluation includes measurement of urinary GAG concentration Definitive diagnosis requires assay of enzyme activity, usually in peripheral blood leukocytes Supportive management can improve the quality of life for affected individuals and their families ERT,HSCT reduce the progression of somatic involvement but not neurological involvement. Summary Ross CJ, Bastedo L, Maier SA, Sands MS, Chang PL. Treatment of a lysosomal storage disease, mucopolysaccharidosis VII, with microencapsulated recombinant cells. Hum Gene Ther. Oct 10 2000;11(15):211727