Osteogenesis Imperfecta
•Download as PPTX, PDF•
127 likes•35,888 views

Osteogenesis imperfecta (OI), also known as brittle bone disease, is a genetic disorder characterized by bones that break easily. It is caused by mutations in genes that produce type 1 collagen, which is important for bone strength. Symptoms can include bone fractures, skeletal deformities, weak bones, hearing loss, and blue sclera. Treatment focuses on surgery to repair bones, bracing to prevent deformities, and bisphosphonates to increase bone density and reduce fractures.

Recommended
More Related Content
What's hot
What's hot (20)
Viewers also liked
Viewers also liked (11)
Similar to Osteogenesis Imperfecta
Similar to Osteogenesis Imperfecta (20)
More from Paudel Sushil
More from Paudel Sushil (19)
Recently uploaded
Recently uploaded (20)
Osteogenesis Imperfecta
- 1. • OSTEOGENESIS IMPERFECTA
- 2. Earliest known case of osteogenesis imperfecta in a partially mummified infant’s skeleton from ancient Egypt now housed in the British Museum in London. In 1835, Lobstein coined the term osteogenesis imperfecta Other names for OI: Lobstein disease, brittle- bone disease, blue-sclera syndrome, and fragile-bone disease
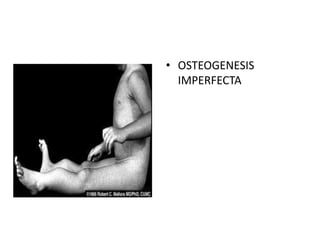
- 3. Manifest itself with 1 or more of the following findings: Blue sclerae Triangular facies Macrocephaly Hearing loss Defective dentition Barrel chest Scoliosis Limb deformities Fractures Joint laxity Growth retardation Constipation and sweating
- 4. Pathologic changes seen in all tissues in which type 1 collagen is an important constituent (eg, bone, ligament, dentin, and sclera) Basic defect : qualitative or quantitative reduction in type 1 collagen Mutations in genes encoding type 1 collagen affect the coding of 1 of the 2 genes Mutations are either genetically inherited or new Inherited mutations : recurrence risk in subsequent pregnancies of 50% if a parent is affected New mutations unpredictable recurrence risk
- 5. Quantitative defects of type 1 collagen : mutations on COL1A gene, production of premature stop codon or a microsense frame shift, which leads to mutant messenger RNA (mRNA) in the nucleus Cytoplasm contains normal alpha1 mRNA; reduced amounts of structurally normal collagen produced Mild form of disease
- 6. Qualitative defects of type 1 collagen: autosomal dominant mutations on either the COL1A or the COL1B gene, production of mixture of normal and mutant collagen chainstype 1 collagen thus formed is functionally impaired because of mutant chain
- 7. In bone :both endochondral and intramembranous ossification affected Epiphysis and physis :broad and irregular, with disorganization of proliferative and hypertrophic zones ,loss of typical columnar arrangement, thinning of zone of calcified cartilage, deficiency of primary spongiosa of the metaphysis and delay of the secondary centers of ossification in the epiphysis.
- 8. Scoliosis and kyphosis Vertebral bodies :wedged, translucent, and shallow
- 9. Thinning of the skull and multiple ossification centers (wormian bones) are present, particularly in the occiput
- 10. Epidemiology Incidence : 1 case for every 20,000 live births Equally common in males and females Described in every human population in which skeletal dysplasias have been studied No predilection for a particular race
- 11. History Family history , but most cases due to new mutations Commonly present with fractures after minor trauma In severe cases, prenatal screening ultrasonography performed during the second trimester may show bowing of long bones, fractures, limb shortening, and decreased skull echogenicity. Lethal OI cannot be diagnosed with certainty in utero
- 12. Physical Examination Clinical presentation depends on phenotype Sillence classificatiom : 4 types on basis of clinical and radiologic features Dentinogenesis imperfecta denoted as subtype B, whereas OI without dentinogenesis imperfecta is denoted as subtype A
- 14. Many cases of OI do not fit into the aforementioned categories; osteoporosis- pseudoglioma, Bruck syndrome, and Cole- Carpenter syndrome. Osteoporosis-pseudoglioma syndrome : caused by mutations in gene encoding for low-density-lipoprotein receptor-related protein 5 (LRP5), with clinical features including blindness and bone fragility
- 15. Bruck syndrome: autosomal recessive condition caused by mutations in bone- specific collagen type 1 telopeptide lysyl hydroxylase enzyme, with clinical features that include congenital joint contractures and bone fragility Cole-Carpenter syndrome : severe progressive form of OI, with associated multisutural craniosynostosis and growth failure
- 16. Complications Repeated respiratory infections Basilar impression caused by a large head, which causes brainstem compression Cerebral hemorrhage caused by birth trauma High risk for complications of anesthesia
- 17. Diagnostic Considerations Differential diagnoses categorized into 3 stages of life: Prenatal/neonatal Preschool/childhood Adolescence/adultho od
- 18. Conditions that should be considered in prenatal/neonatal stage include: Jeune dystrophy Camptomelic dysplasia Chondrodysplasia punctata Chondroectodermal dysplasia (Ellis–van Creveld syndrome) Nonaccidental injury Hypophosphatasia
- 19. Preschool/childhood stage include: Pyknodysostosis Hajdu-Cheney syndrome Osteochondromatosis Nonaccidental injury
- 20. Differentiate between OI and child abuse Keys to distinguishing OI from child : Metaphyseal corner fractures, which are common in child abuse, rare in OI In children with OI, fractures may continue to occur while they are in protective custody Child abuse has nonskeletal manifestations (eg, retinal hemorrhage, visceral intramural hematomas, intracranial bleeds of various ages, pancreatitis, and splenic trauma)
- 21. Differential Diagnoses Achondroplasia Menkes Kinky Hair Disease Glucocorticoid Therapy Cushing Syndrome Homocysteinemia McCune-Albright Syndrome Osteopetrosis Osteoporosis Pediatric Acute Lymphoblastic Leukemia Rickets Scurvy Thanatophoric Dysplasia Wilson Disease
- 22. Laboratory Studies Within reference ranges, and useful in ruling out other metabolic bone diseases An analysis of type I, III, and V collagens synthesized by fibroblasts helpful Collagen synthesis analysis : culturing dermal fibroblasts obtained during skin biopsy Results are negative in syndromes resembling OI.
- 23. Tests Sodium dodecyl sulfate– polyacrylamide gel electrophoresis (SDS- PAGE) 2-Dimensional SDS-PAGE Cyanogen bromide (CNBr) mapping Thermal stability studies An analysis of amino acid composition of collagens
- 24. DNA blood testing for gene defects has an accuracy of 60- 94%. Prenatal DNA mutation analysis can be performed in pregnancies with risk of OI to analyze uncultured chorionic villus cells. Samples are obtained during chorionic villus sampling performed under ultrasonographic guidance when a mutation in another member of the family is already known
- 25. Prenatal ultrasonography : Useful in evaluating OI types II and III Detects limb-length abnormalities at 15-18 weeks Features include supervisualization of intracranial contents caused by decreased mineralization of calvaria (also calvarial compressibility), bowing of the long bones, decreased bone length (especially of the femur), and multiple rib fractures
- 26. Radiographic skeletal survey after birth Plain radiographs :3 radiologic categories of OI A. Category I – Thin and gracile bones B. Category II – Short and thick limbs C. Category III – Cystic changes
- 27. Radiologic features Fractures – Commonly, transverse fractures and those affecting the lower limbs Excessive callus formation and popcorn bones - Multiple scalloped, radiolucent areas with radiodense rims Skull changes - Wormian bones enlargement of frontal and mastoid sinuses, and platybasia with or without basilar impression Deformities of the thoracic cage - Fractured and beaded ribs and pectus carinatum Pelvic and proximal femoral changes - Narrow pelvis, compression fractures, protrusio acetabuli, and shepherd’s- crook deformities of the femurs
- 28. Mild OI (type I) : thinning of the long bones with thin cortices,wormian bones,no deformity of long bones Extremely severe OI (type II) : beaded ribs, broad bones, and numerous fractures with deformities of long bones Moderate and severe OI (types III and IV) :cystic metaphyses, or a popcorn appearance of growth cartilage, deformities of long bones, old rib fractures, vertebral fractures
- 30. Dual x-ray absorptiometry (DEXA) To assess bone mineral density in children with milder forms Bone mineral density low in children and adults regardless of severity. Bone mineral densities can be normal in infants with OI, even in severe cases In pediatric patients, DEXA results not useful for predicting risk of fracture No reliable published reference data regarding DEXA in infants available
- 31. Polarized light microscopy or microradiography used in combination with scanning electron microscopy to assess dentinogenesis imperfecta With skin biopsy, collagen can be isolated from cultured fibroblasts and assessed for defects, with an accuracy of 85-87% Bone biopsy : show changes in concentrations of noncollagenous bone proteins, such as osteonectin, sialoprotein, and decorin
- 32. Histologic Findings • Width of biopsy cores, width of cortex, and volume of cancellous bone decreased in all types of OI • Number and thickness of trabeculae reduced • Evidence of defects in modeling of external bone in terms of size and shape production of secondary trabeculae by endochondral ossification, thickening of secondary trabeculae by remodeling
- 33. Treatment No cure Orthotics: limited role, to stabilize lax joints (eg, ankle and subtalar joints with ankle-foot orthoses) and to prevent progressive deformities and fractures. Provide walking aids, specialized wheelchairs, and home adaptation devices to help improve patient’s mobility and function
- 34. Surgery Pillar of treatment Only if it is likely to improve function and treatment goals are clear Intramedullary rod placement, surgery to manage basilar impression, and correction of scoliosis Soft tissue surgery : lower-limb contractures, particularly those of the Achilles tendon
- 35. Painful bony deformities and recurrent fractures are typically treated with intramedullary stabilization with or without corrective osteotomies. In children with severe forms of OI (eg, type III), rodding of lower extremities is performed to correct deformities and provide preventive protection around the time of first attempts at standing Because bone is soft in OI, rods (eg, extendable Sheffield rods or Bailey-Dubow rods), pins (eg, Rush pins), and wires (eg, Kirschner wires) are used rather than solid nails, plates, and screws; the latter are associated with increased fracture risk above and below the device and with poor fixation
- 36. Rod placement use in femur and less commonly used in tibia, humerus, and forearm In the prebisphosphonate era, extendable rods preferred to nonextendable ones in order to prevent bone bowing and bone growth beyond end of rod Bailey-Dubow rods : high incidence of mechanical failures (eg, migration and disconnection of T-parts) Sheffield rods and the Fassier-Duval modification commonly used
- 37. With decreased fragility of bone exposed to bisphosphonate, future role of extendable rods unclear In long bones (eg, tibiae and radii), nonextendable rods such as Rush pins and Kirschner wires most often used Complications of rod placement include breakage, rotational deformities, and migration Extendable and nonextendable rods associated with similar complications Rate of repeat surgical intervention is lower with extendable rods than with nonextendable rods
- 38. Surgery for basilar impression Basilar invagination: result in long tract signs and respiratory depression from direct compression of brainstem and upper cervical and cranial nerves Treated with decompression and stabilization of the craniocervical junction; reserved for cases with neurologic deficiencies
- 39. Surgery for spinal deformities Bracing not effective in treating spinal deformities such as scoliosis and kyphosis, because the rib cage is fragile to transfer brace pressure to vertebral column. External pressure may worsen the chest deformities. Surgery is indicated when the following 2 conditions are present: Acceptable bone quality Progressive scoliosis with curvature of more than 45° if OI is mild or more than 30-35° if OI is severe
- 40. Posterior spinal arthrodesis is the treatment of choice and is best performed with segmental instrumentation. Often, significant correction and stable fixation are not achieved. Pretreatment with pamidronate appears to improve the surgical outcome
- 41. Skilled administration of anesthetics and awareness of the limitations of surgery are essential prerequisites. Anesthetic-related problems : Patients with relatively large heads and tongues and in those with short necks Chest deformities may cause respiratory complications On the operating table, fractures may arise as a result of the application of a blood pressure cuff or tourniquet, or they may occur during transfers Watch for hyperthermia and increased sweating
- 42. Bisphosphonates Synthetic analogues of pyrophosphate that inhibit osteoclast- mediated bone resorption on the endosteal surface of bone by binding to hydroxyapatite. Unopposed osteoblastic new bone formation on the periosteal surface results in an increase in cortical thickness.
- 43. Cyclic intravenous (IV) pamidronate : Dosage of 7.5 mg/kg/y at 4- to 6-month intervals Dosages have ranged from 4.5 to 9 mg/kg/y, depending on the protocol used Cyclic administration of IV pamidronate reduces the incidence of fracture and increases bone mineral density Current evidence does not support the use of oral bisphosphonates in patients with OI. IV pamidronate effective in babies and can be used to relieve pain in severe cases Adverse effects of pamidronate : acute febrile reaction, mild hypocalcemia, leukopenia, a transient increase in bone pain, and scleritis with or without anterior uveitis
- 44. Risedronate, alendronate, and zoledronic acid being assessed Growth hormone: act on growth plate,stimulate osteoblast function, possibly via IGF-1 ,IGFBP-3 Teriparatide : Recombinant human form of parathyroid hormone that increases number and activity of osteoblasts Potential use of teriparatide for the treatment of OI remains to be defined
- 45. Cellular and Genetic Therapy Bone marrow transplantation: potential future therapeutic modality for OI Because there are very few MSCs in the average human bone marrow graft, approaches involving expansion of the number of MSCs in ex vivo cultures with subsequent infusion into the recipient needed Such cell therapies usually result in somatic mosaicism, where normal and abnormal osteoblasts exist in the same body Unfortunately, higher proportion of engrafted normal cells required to achieve the level of normal osteoblasts necessary to functionally correct the OI phenotype. Use of immunosuppressive agents to prevent graft rejection and graft versus host reaction can itself damage bone
- 46. • Future approaches: autografting of genetically modified mutant osteoblasts, whereby mutant collagen gene is inactivated • Gene therapy: being explored in animal models, but major obstacles remain, both because of intrinsic difficulties and because of dominant negative mechanism of disease
- 47. Diet and Activity Nutritional evaluation and intervention paramount to ensure appropriate intake of calcium, phosphorus, and vitamin D Caloric management important, particularly in adolescents and adults with severe forms of OI Physical therapy, in form of comprehensive rehabilitation programs, directed toward improving joint mobility and developing muscle strength
- 48. In early infancy, gentle handling of babies by parents to prevent fractures, with frequent positional changes advised to prevent occipital flattening, torticollis, and frog-leg positioning of hips When infant is crawling: upper-limb mobility, propelling a wheelchair or ambulating with walking aids When child starts to stand: walking encouraged, both as exercise and as primary or secondary means of mobility
- 49. Weightbearing promoted in pool, on tricycles, and with walkers Prone positioning to prevent hip flexion contractures; aided by strengthening of hip extensors and quadriceps. Bisphosphonates have significantly improved the walking ability of children with severe forms of OI
- 50. Care of patients with OI multidisciplinary: occupational therapist, physical therapist, nutritionist, audiologist, orthopedic surgeon, neurosurgeon, pneumologist, and nephrologist, among others Genetic counseling to parents of child with OI who plan to have more children
- 51. Prognosis Morbidity and mortality vary widely, depending on genotype Variability occurs between individuals with different mutations Life expectancy of subjects with nonlethal OI appears same as that for the healthy population, except for those with severe respiratory or neurologic complications. Although patients with lethal OI may die in perinatal period, individuals with extremely severe OI can survive until adulthood
- 52. Patient Education Patients with OI: well motivated and keen to achieve as much as possible despite their physical limitations Education extremely important Education of parents and families :to know how to position child in crib and how to hold child so as to minimize risk of fractures while maintaining bonding and physical stimulation
- 53. Living with ostogenesis imperfecta The tips reproduced below have been developed by the Osteogenesis Imperfecta Foundation for taking care of children with osteogenesis imperfecta. Do not be afraid to touch or hold an infant with osteogenesis imperfecta, but be careful. To lift an infant with osteogenesis imperfecta, spread your fingers apart and put one hand between the legs and under the buttocks, and place the other hand behind the shoulders, neck, and head. Never lift a child with osteogenesis imperfecta by holding him or her under the armpits.
- 54. Do not pull on arms or legs or, in those with severe osteogenesis imperfecta, lift the legs by the ankles to change a diaper. Select an infant car seat that reclines. It should be easy to place or remove your child in the seat. Consider padding the seat with foam and using a layer of foam between your child and the harness. Be sure your stroller is large enough to accommodate casts. Do not use a sling- or umbrella-type stroller
- 55. Follow your doctor's instructions carefully, especially with regard to cast care and mobility exercises. Swimming and walking are often recommended as safe exercises. Adults with osteogenesis imperfecta should avoid activities such as smoking, drinking, and taking steroids because they have a negative impact on bone density. Increasing awareness of child abuse and a lack of awareness about osteogenesis imperfecta may lead to inaccurate conclusions about a family situation. Always have a letter from your family doctor and a copy of your child's medical records handy.