ICH Q11 DEVELOPMENTAND
MANUFACTURE OF DRUG
SUBSTANCES
ICH Q11 complements Q8 (development), Q9 (risk
management) and Q10 (pharmaceutical quality
system), but focuses specifically on drug substance
development.
2.
Introduction, Scope andTarget
• Harmonization of scientific and technical principles relevant to the design,
development & manufacture of drug substances as part of a total control
strategy designed to ensure product quality & consistency.
• Provide guidance on the information to be provided in CTD Sections
3.2.S.2.2 – 3.2.S.2.6
• Provide further clarification on principles & concepts described in ICH
guidelines Q6,Q8, Q9 & Q10 as they pertain to the specification,
development and manufacture of drug substance.
• To establish a commercial manufacturing process capable of consistently
producing drug substance of the intended quality.
3.
Manufacturing Process Development
Generalprinciples
• Drug substance quality link to drug product
• Process development tools
• Approaches to development
• Drug substance critical quality attributes (CQAs)
• Linking material attributes & process parameters to drug substance CQAs
• Design space
Submission of information
• Overall process development summary
• Drug substance CQAs
• Manufacturing process history
• Manufacturing developmental studies
4.
Process Development Tools
•Quality Risk Management (as described in ICH Q9)
• Assessing quality attributes & manufacturing process parameters.
• Increasing assurance of routine achievement of acceptable results.
• Risk assessment can be carried out early in the development and shall be
repeated as knowledge & understanding increases on the process.
• Knowledge Management (as described in ICH Q10)
• Prior knowledge & development studies which include chemical, engineering
principles and applied manufacturing experience.
These tools help establish the link between material attributes, process
parameters, and drug substance quality.
5.
Approaches to Development
•Approaches of development vary from company to company & product to product.
Traditional Approach:
• Identifying potential CQAs having impact on product quality.
• Defining appropriate manufacturing process.
• Defining a control strategy to ensure process performance & quality.
Enhanced Approach:
• Prior knowledge, experimentation and risk assessment.
• Determining linkage between material attributes & process parameters to CQAs.
• Using enhanced approach in combination with Quality Risk Management to establish
appropriate control strategy.
• Traditional & Enhanced approaches are not mutually exclusive.
• In real practice, most companies apply a hybrid of traditional and enhanced
approaches.
6.
Drug substance CriticalQuality Attributes (CQAs)
• CQA is a physical / chemical / biological / microbiological property that should be
within an acceptance criteria to ensure the desired product quality.
• CQAs are used to guide process development.
• CQAs can be modified as the knowledge & process understanding increases.
• CQAs include the properties that can affect identity, purity, biological activity and
stability.
• Impurities are important aspect of drug substance CQAs due to their potential
impact on drug product safety.
7.
Example: Linking MaterialAttributes and Process
Parameters to Drug Substance CQAs - Chemical Entity
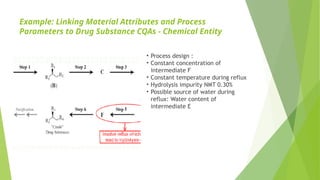
• Process design :
• Constant concentration of
intermediate F
• Constant temperature during reflux
• Hydrolysis impurity NMT 0.30%
• Possible source of water during
reflux: Water content of
intermediate E
8.
• Based onrisk assessment, parameters affecting hydrolysis :
• Time of reflux &
• Water content of intermediate E
• The reaction was expected to follow second-order kinetics according to the
equation below:
9.
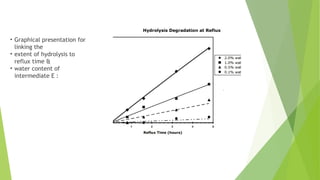
• Graphical presentationfor
linking the
• extent of hydrolysis to
reflux time &
• water content of
intermediate E :
10.
• Control strategyby Traditional approach :
Dry intermediate E to a maximum water content of 1.0%
Target -reflux time to 1.5 hours & maximum of 4 hours
• Control strategy by Enhanced approach :
2nd order rate equation can be integrated (Chemical Reaction Engineering,
Levenspiel 2nd Edition, 1972)
11.
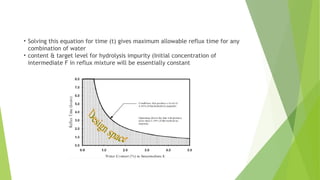
• Solving thisequation for time (t) gives maximum allowable reflux time for any
combination of water
• content & target level for hydrolysis impurity (Initial concentration of
intermediate F in reflux mixture will be essentially constant
12.
Description of manufacturingprocess
and process controls
• Manufacturing process should be provided as follows,
• Process flow diagram,
• Sequential procedural narrative,
• In-process controls to be indicated in the process description,
• Scaling factors, if the process is scale dependant,
• Should include design space (if any).
13.
Selection of startingmaterials and
source materials
General principles
• Selection of starting materials for synthetic drug substances
• Selection of starting materials for semi-synthetic drug substances
• Selection of source materials for Biotechnological / Biological products
Submission of information
• Justification of starting materials selection for synthetic drug substances
• Justification of starting materials selection for semi-synthetic drug Substances
• Qualification of source materials for Biotechnological / Biological products
14.
Control Strategy
• Controlstrategy is a planned set of controls, derived from product and process
understanding, that assures process performance & quality. It include,
Control on material attributes
Controls implicit in the design of manufacturing process
In-process controls
Controls on drug substance
15.
Process Validation /Evaluation
• Process validation involves the collection and evaluation of data, from the
process design stage throughout production, that establish scientific evidence
that a process is capable of consistently delivering a quality drug substance.
Process should be validated (ICH Q7) before commercial distribution.
Number of batches to be considered for validation depends on
The complexity of process,
Level of process variability,
Amount of experimental data / process knowledge available on specific
process.
For non-sterile drug substances, results of process validation are not normally
included in the dossier.
16.
SUBMISSION OF MANUFACTURING
PROCESSDEVELOPMENT & RELATED
INFORMATION IN CTD FORMAT
Quality risk management and process development in 3.2.S.2.6.
Critical Quality Attributes in 3.2.S.2.6 & 3.2.S.3.1, 3.2.S.4.1 & 3.2.S.7 (if relevant).
Design space in 3.2.S.2.2, 3.2.S.2.4, 3.2.S.2.6, 3.2.S.4.5 (wherever the relevant
justification is applicable).
Control strategy in 3.2.S.4.5 and can also be in 3.2.S.2.2, 3.2.S.2.4, 3.2.S.2.6,
3.2.S.4.1.
17.
Life Cycle Management
Product & process knowledge should be managed from development through
the commercial life of the product up to discontinuation.
Process performance, control strategy & suitability of design spaces should be
periodically evaluated through out the life cycle and can be done as a part of
Product Quality Review (ICH Q7).
It includes process development, technology transfer, process validation and
change management activities.
Any changes within the design space (if submitted in DMF) does not require any
approval by regional regulatory authorities.
There should be a systematic managing knowledge related to both drug
substance & manufacturing process throughout the life cycle.