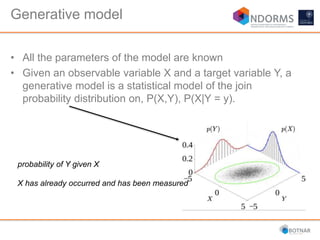
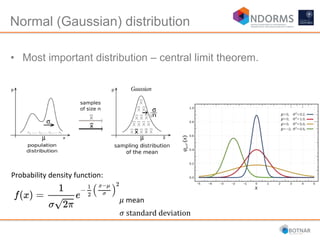
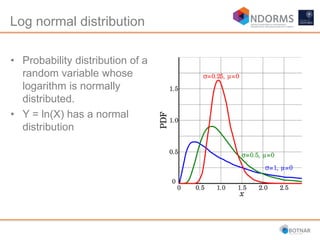
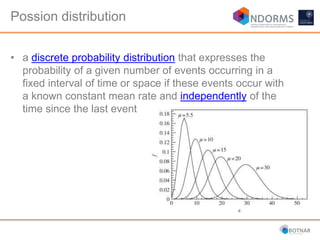
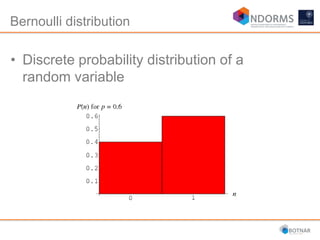
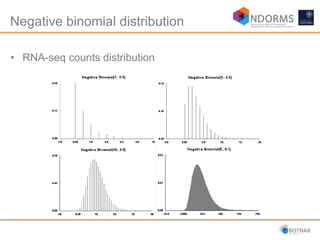
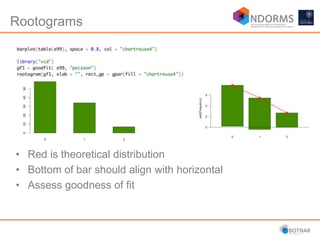
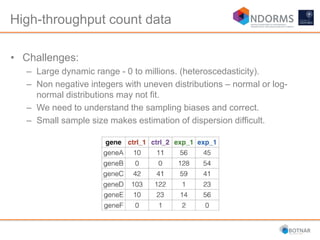
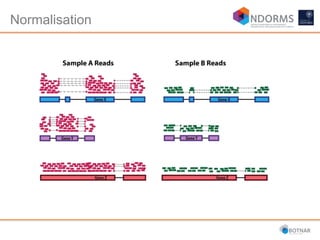
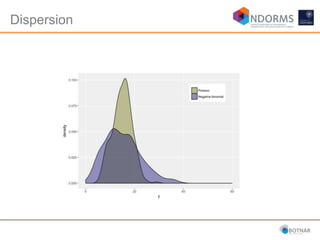
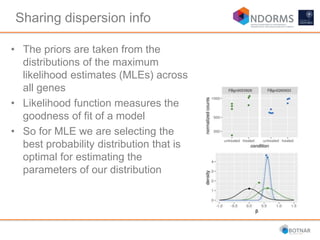
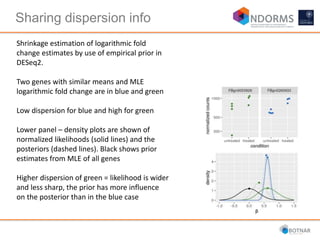
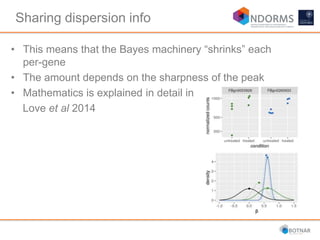
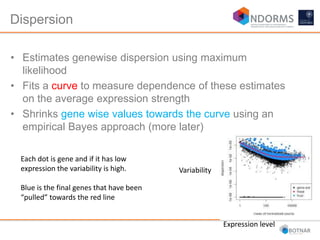
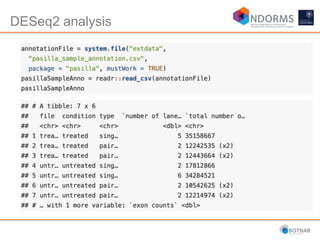
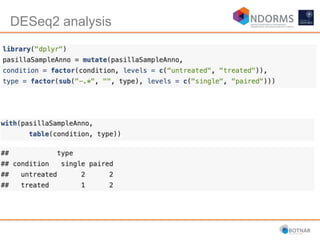
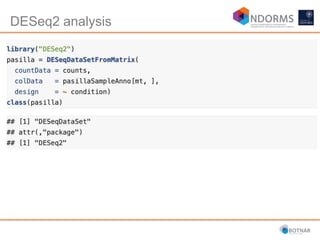
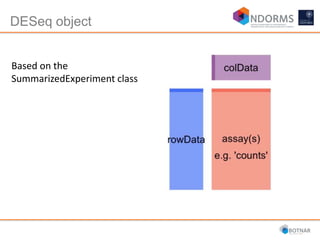
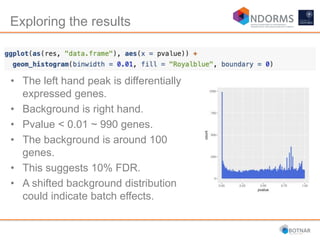
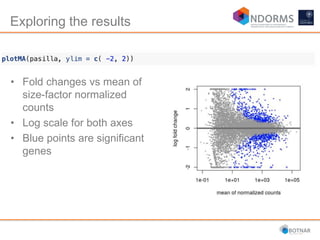
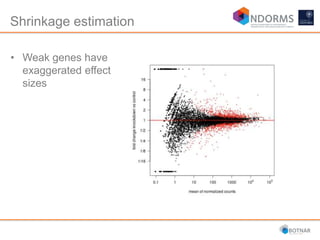
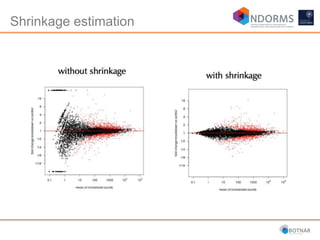
DESeq2 is used to analyze differential expression from RNA-seq count data using a generalized linear model. It models counts using a gamma-Poisson distribution and estimates dispersion using empirical Bayes shrinkage. Key steps include normalizing counts, estimating dispersion, fitting the linear model, and using Wald and likelihood ratio tests to identify differentially expressed genes while controlling the false discovery rate. Results can be explored using plots of p-values, mean-variance trends, ordination plots, and heatmaps to visualize sample relationships and differentially expressed genes.