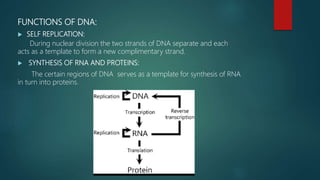
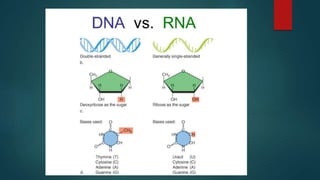
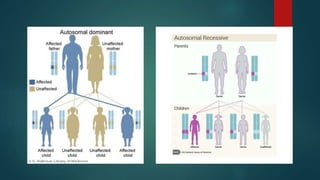
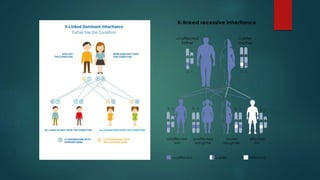
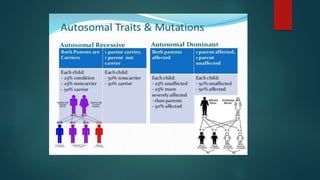
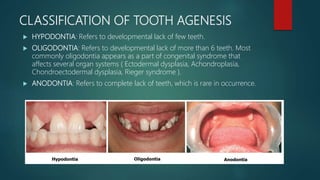
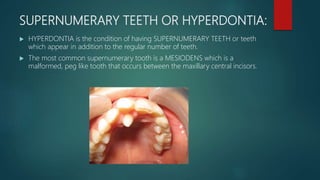
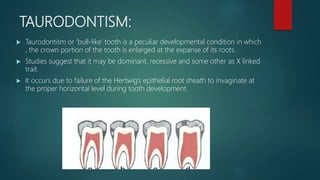
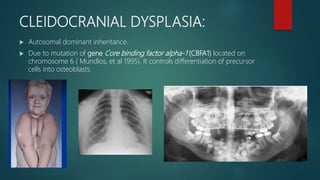
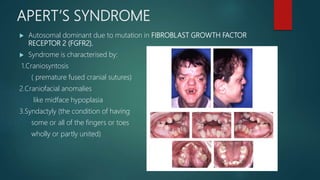
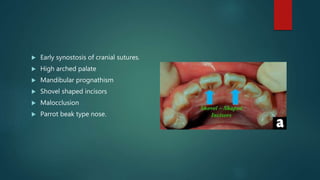
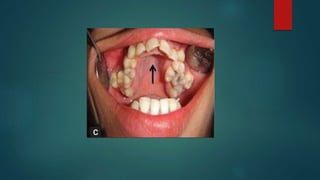
The document presents an extensive overview of genetics, detailing definitions of key terms such as genes, chromosomes, and various genetic disorders related to dental development. It also discusses the role of specific genes in tooth formation and related anomalies, including tooth agenesis and hyperdontia, alongside their inheritance patterns. Additionally, it covers the structure and function of DNA and RNA, the genetic basis for certain developmental disorders affecting teeth, and introduces complex concepts such as X chromosome inactivation and related hypotheses.





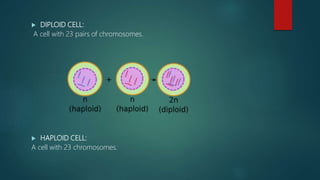
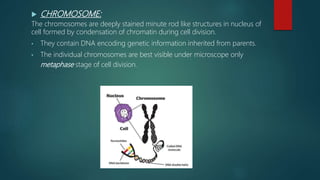
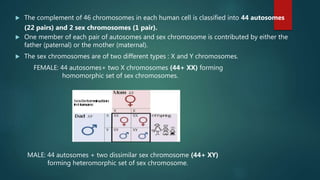








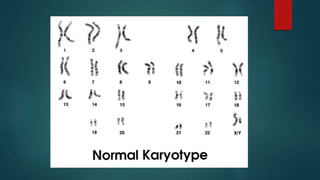


















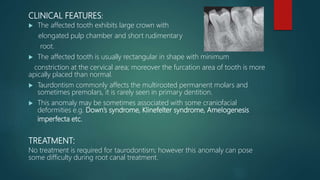
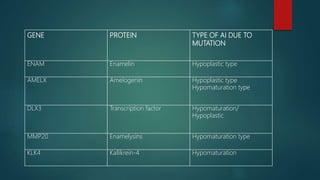

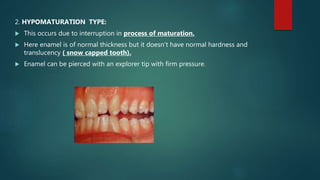






















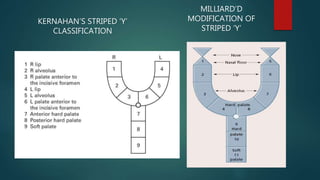





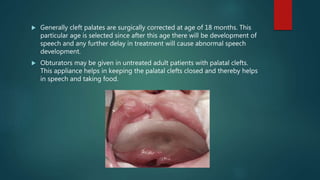


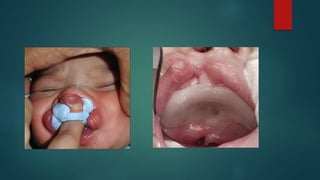








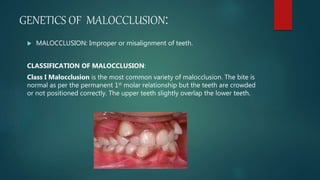
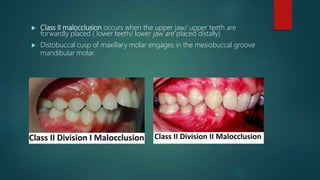
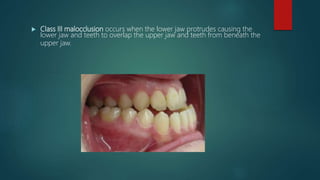
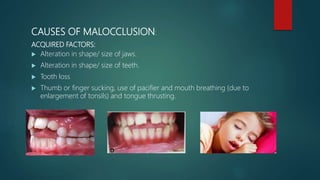










































![Introduction to Genetics[physiology] 2024.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/genetics2024-240627074103-782b01a0-thumbnail.jpg?width=640&height=640&fit=bounds)



































