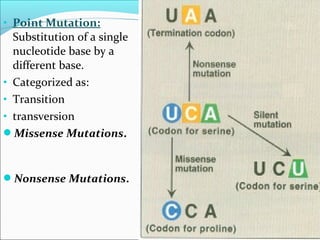
Genetics is the study of heredity and genetic variation. Key terms include:
- Genotype is the genetic makeup of an organism, phenotype is observable traits.
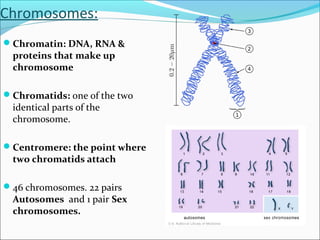
- Genes hold information to build cells and pass traits to offspring. The human genome contains 25,000-35,000 genes located on 23 chromosome pairs in the nucleus.
- DNA is transcribed to RNA and translated to proteins, which determine an organism's traits. Variations in genes and chromosomes can result in genetic disorders. Common methods to study genetics include karyotyping, analyzing pedigrees, and identifying alleles and mutations. Understanding genetics provides insight into inheritance patterns and human health.


























![ALLELE
A variant of the DNA sequence at a given locus is
called an allele
Homozygous-an organism in which 2 copies of genes are
identical i.e. have same alleles [AA/ aa]
Heterozygous-an organism which has different alleles of
the gene [Aa]
Do minant alleles are represented with a capitalcapital letter
Recessive alleles are represented with a lo wer caselo wer case letter
Mutation can cause variation in alleles.](https://image.slidesharecdn.com/geneticsppt-180508182109/85/Genetics-28-320.jpg)