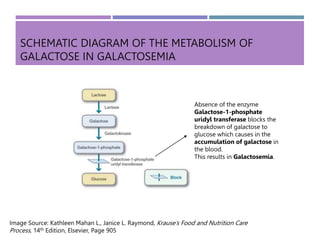
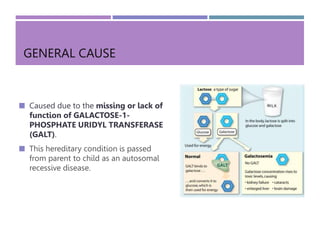
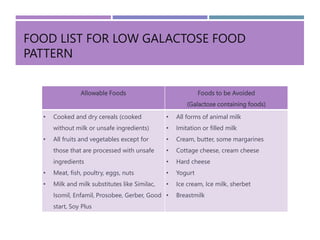
Galactosemia is a genetic disorder characterized by the inability to properly metabolize the sugar galactose, leading to its accumulation in the blood. It is caused by a deficiency of the enzyme galactose-1-phosphate uridyl transferase and exhibits various symptoms and complications, including cognitive and developmental issues. Treatment involves strict galactose restriction through medical nutrition therapy to manage the disorder's effects and prevent complications.





















































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)



![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)

