Sequence Alignment
Significance ofSequence Alignment
Methods of Sequence Alignment
Dot Plot
Principle
Dot Plot Algorithm
Examples and interpretations of dot plots
Dot Plot Software
Application
Limitation
TABLE OF CONTENT:
3.
Why Sequence Alignment?
Sequence Alignment is the procedure of comparing
Two (Pair-wise Alignment) or More (Multiple
sequence Alignment) sequences by searching for a
series of individual characters or character patterns that
are in the same order in the sequences.
It is an Important First Step toward Structural and
Functional Analysis of newly Determined Sequence .
Sequence Alignment
Global
Alignment
Local
Alignment
In Global alignment,two sequences to be aligned over their
entire length.
Sequences that are quite similar and approximately Have
the same length are suitable for global alignment.
The alignment is performed from start to finish of the two
sequences in order to find the best possible alignment
GLOBALALIGNMENT :
Taking Entire Part of the Sequence
6.
LOCALALIGNMENT :
Itonly finds local regions with the highest level of
similarity between the two sequences and aligns
these regions without regard for the alignment of the
rest of the sequence regions.
Find the Best Matching subsequence
7.
Significance of SequenceAlignment:
Sequence alignment is useful for discovering Functional,
Structural, and Evolutionary information in biological
sequences
Functional: DNA molecules that are very much alike or similar
in sequence analysis parlance probably have the same
regulatory role.- Protein molecules that are very much alike
probably have the same biochemical function
Structural: Protein molecules that are very much alike
probably have the same 3-D structure
Evolutionary: If two sequences from different organisms are
similar then there may have been a common ancestor
sequence, and the sequences are then defined as being
homologous .The alignment indicates the changes that could
have occurred between the two homologous sequences and a
common ancestor sequence during evolution.
8.
Methods Of SequenceAlignment :
Alignment of Pairs of Sequences:
Alignment of two sequences is performed using
the following methods:
Dot matrix analysis
The dynamic programming (or DP)
algorithms
Word or k-tuple methods, such as used by the
programs FASTA and BLAST,
9.
DOT PLOT :
InBioinformatics a dot plot is a graphical method that
allows the comparison of two biological sequences and
identify regions of close similarity between Them
Method for comparing two amino acid or nucleotide
sequences
Established in 1970 by A.J. Gibbs and G. A. McIntyre
Dot plots are useful as a first-level filter for determining
an alignment between two sequences.
It reveals the presence of insertions or deletions
10.
DOT PLOT :
Principle:
Dot plot are two dimensional graphs, showing a Comparison Between of two sequences. The
principle used to generate the dot plot is: The top X and the left y axes of a rectangular array
are used to represent the two sequences to be compared
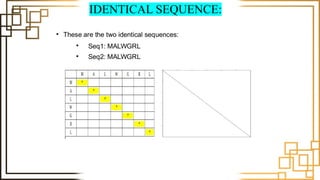
Example:
Seq 1: GCTGAA
Seq 2: GCGAA
G C T G A A
G
C
T
A
A
11.
DOT PLOT :
DotPlot Algorithm :
A dot plot is a visual representation of the similarities between two sequences.
One sequence (A) is listed across the top of the matrix and the other (B) is listed
down the left side
Starting from the first character in B, one moves across the page keeping in the
first row and placing a dot in many column where the character in A is the same B
The process is continued until all possible comparisons between A and B are made
Any region of similarity is revealed by a Diagonal row of dots
Isolated dots not on diagonal represent random matches.
12.
DOT PLOT ALGORITHM:
Example:
Seq 1: TWILIGHTZONE
Seq 2: MIDNIGHTZONE
T W I L I G H T Z O N E
M
I
D
N
I
G
H
T
Z
O
N
Calculation: Matrix
• Rows = residues of sequence 1
• Column= residues of sequence 2.
A dot is plotted at every co-ordinate
where there is similarity between the
bases
FRAME SHIFTS MUTATION:
Whatis frameshift mutation?
A Frameshift mutation (also called a framing error or a reading frame
shift) is a genetic mutation caused by indels (insertions or deletions) of a
number of nucleotides in a DNA sequence
20.
Window Size :
DOTPLOT BECOMES TOO NOISY WHEN WE COMPARE LARGE AND SIMILAR
SEQUENCES:
By using sliding window (size =w), cut-off (value=v)
A residue by residue comparison (window size = 1) would undoubtedly result in a
very noisy background due to a lot of similarities between the two sequences of
interest. For DNA sequences the background noise will be even more dominant
as a match between only four nucleotide is very likely to happen. Moreover, a
residue by residue comparison (window size = 1) can be very time consuming and
computationally demanding.
So Increase the Window Size
Window size changes with goal of
analysis
– size of average exon
– size of average protein structural
element
– size of gene promoter
– size of enzyme active site
21.
Dot Plot Software:
GCGis a commercial software, hence not possible to use all the time.
Instead of this, we can use the EMBOSS package, which are followig:
Dotmatcher
Dotpath
Polydot
Dottup
22.
Shows theall possible alignment between two nucleic acid and amino acid sequences.
All kind of local and global alignment can be traped .
To find self base pairing of RNA (eg, tRNA) by comparing a sequence to itself
complemented and reverse.
An excellent approach for finding sequence transposition.
To find the location of genes between two genomes.
Dot plot applications are particularly useful in the identification of interspersed repeats
such as transposons and tandem-repeat motifs such as microsatellites.
Furthermore, loss or gain of whole motifs can easily be spotted in different types of
domains, a trait useful in characterising the evolution of certain protein families.
Dot plots are also employed in the investigation of properties of protein coding
sequences by predicting secondary structures, like stem-loop formation or structural
RNA domains.
Can use to find self base pairing of an RNA (e.g., tRNA) by comparing a sequence to
‐
itself complemented and reversed.
APPLICATION :
LIMITATION :
Forlonger sequence, memory required for the graphical
representation is very high.
So long sequence can not be aligned.
Lots of Insignificant matches makes it noisy (so many
off diagonal appear).
Time required to compare two sequences is proportional
to the product of length of the sequences time of the
search window.
i.e, Higher efficiency of short sequence.
Low efficiency of long sequence.






















![Out put
Tubulin-specific
chaperone
A
isoform
2
[Homo
sapiens]
Tubulin-specific chaperone A
Oryctolagus cuniculus (Rabbit)](https://image.slidesharecdn.com/dotplot-250823113513-5654f11e/85/Dot-Plot-in-statistics-applications-pptx-24-320.jpg)








































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)










