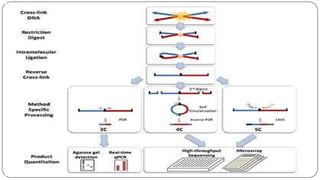
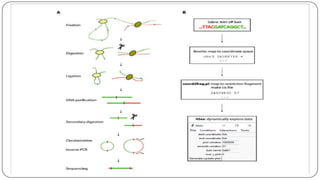
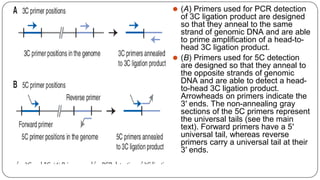
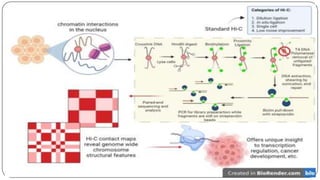
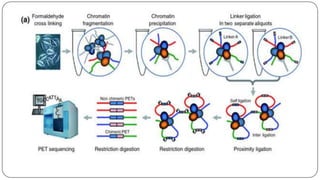
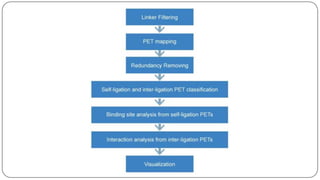
The document discusses the significance of DNA-protein interactions in gene regulation and genome stability, detailing various techniques like 3C, 4C, 5C, Hi-C, and ChIA-PET that study these interactions. It emphasizes the advancements in studying the three-dimensional genome organization and how these methods can analyze chromatin interactions at high resolution. Additionally, it outlines the methodologies and limitations of each technique in understanding DNA topology and gene regulation.