Hypertensive Heart Disease and Cardiomyopathies
•Download as PPT, PDF•
13 likes•3,509 views

This document discusses hypertensive heart disease and cardiomyopathies. It describes how hypertensive heart disease can lead to left ventricular hypertrophy over time as an adaptive response to chronic hypertension. If compensated, it may be asymptomatic, but can eventually lead to heart failure, arrhythmias or sudden death. Three main types of cardiomyopathy are discussed - dilated, hypertrophic and restrictive cardiomyopathy. Dilated cardiomyopathy is characterized by ventricular dilation and contractile dysfunction, while hypertrophic cardiomyopathy involves thickened heart walls and restrictive cardiomyopathy decreases ventricular compliance. Various genetic and acquired causes are provided for each type.

Recommended
More Related Content
What's hot
What's hot (20)
Similar to Hypertensive Heart Disease and Cardiomyopathies
Similar to Hypertensive Heart Disease and Cardiomyopathies (20)
More from Prasad CSBR
More from Prasad CSBR (20)
Recently uploaded
Recently uploaded (20)
Hypertensive Heart Disease and Cardiomyopathies
- 1. Hypertensive Heart DiseaseHypertensive Heart Disease CSBR.Prasad, MD.,CSBR.Prasad, MD., May-2015-CSBRP 1
- 2. • Hypertrophy • Causes for LVH • Causes for RVH • Complications of systemic HTN • Name some causes for cardimegaly? May-2015-CSBRP 2
- 3. Systemic (Left-Sided) Hypertensive Heart Disease • Hypertrophy of the heart is an adaptive response to the pressure overload of chronic hypertension • In the course of time, compensatory changes may be ultimately maladaptive and can lead to myocardial dysfunction, cardiac dilation, CHF or sudden death May-2015-CSBRP 3
- 4. Systemic Hypertensive Heart Disease The minimal pathologic criteria for the diagnosis of systemic HHD are: 1. Left ventricular hypertrophy and 2. Pathologic evidence of hypertension in other organs (e.g., kidney) Even mild hypertension (levels only slightly above 140/90 mm Hg)—if sufficiently prolonged—induces left ventricular hypertrophy May-2015-CSBRP 4
- 5. Systemic Hypertensive Heart Disease Hypertension induces: • Left ventricular hypertrophy – Initially concentric hypertrophy – Left ventricular wall thickening >2cm – Increase in weight of the heart >500gm • Left atrial enlargement (due to diastolic filling) Microscopically: – Increase in transverse diameter of the cardiac muscle fiber – Interstitial fibrosis May-2015-CSBRP 5
- 7. Systemic Hypertensive Heart Disease • Asymptomatic, if compensated – ECG / Echo evidence of LVH • Many patients present with – Atrial fibrillation – CCF Complications: – IHD – Stroke – Renal damage – Progressive cardiac failure • Effective control of HTN reduces the chances of complications May-2015-CSBRP 7
- 8. Pulmonary (Right-Sided) Hypertensive Heart Disease (Cor Pulmonale) • Chronic cor pulmonale is characterized by: – RVH – Right sided heart failure • Acute cor pulmonale can follow massive pulmonary embolism MORPHOLOGY: Acute CP - marked dilation of the RV Chronic CP RVH =/>1cm (Normal thickness 0.3-0.5cm) Thickening of moderator band RVH may encroach on to left ventricle and cause fibrous thickening of Tricuspid valve and TR May-2015-CSBRP 8
- 10. Pulmonary embolism (PE) • The most common ECG finding in the PE is sinus tachycardia • However, the "S1Q3T3" pattern of acute cor pulmonale is classic • This is termed the McGinn-White sign May-2015-CSBRP 11
- 11. Pulmonary (Right-Sided) Hypertensive Heart Disease (Cor Pulmonale) It should also be remembered that PHT most commonly occurs as a complication of left-sided heart disease May-2015-CSBRP 12
- 14. Cardiomyopathies • They are primary diseases of the myocardium • due to genetic causes • Associated with inappropriate ventricular hypertrophy or dilatation • Causing mechanical and/or electrical dysfunction • often leading to cardiovascular death or progressive heart failure May-2015-CSBRP 15
- 15. Cardiomyopathies Presentations: • CCH • Arrhythmias Types: • Primary cardiomyopathies (genetic or acquired diseases of myocardium) • Secondary cardiomyopathies (myocardial involvement as a component of a systemic or multiorgan disorder) May-2015-CSBRP 17
- 16. Exclusion Ventricular dysfunction due to: • Ischemia • Valvular Abnormalities • Hypertension should not be denoted as cardiomyopathies May-2015-CSBRP 18
- 17. ClassificationClassification Can be classified according to a variety of criteria: 1.Underlying genetic basis 2.Arrhythmia-inducing channelopathies 3.Those producing anatomic abnormalities – Three pathologic patterns: • Dilated cardiomyopathy (including ARVH) [90%] • Hypertrophic cardiomyopathy • Restrictive cardiomyopathy [Rarest] May-2015-CSBRP 19
- 18. Can you name some channelopathies? • Cystic fibrosis • Familial periodic paralysis • Long QT syndrome • Myesthenia gravis • Myotonia congenita May-2015-CSBRP 20
- 19. Three major morphologic patterns of cardiomyopathy Note: Only left heart is shown in the diagram May-2015-CSBRP 21
- 21. Dilated Cardiomyopathy Characterized by: • Dilatation • Hypertrophy • Contractile dysfunction May-2015-CSBRP 23
- 22. DCM - PathogenesisDCM - Pathogenesis • Genetic Influences – 30-40% of cases – Mutations in >20 genes [AD > XR > Mitochondrial] • Myocarditis – Coxsackie B • Alcohol and other toxins – Acetaldehyde, Cobalt, Doxorubicin • Childbirth – Peripartum cardiomyopathy • Iron overload - Hemochromatosis • Supraphysiologic stress - Excess Thyroxin, catecholamines May-2015-CSBRP 24
- 25. Schematic of a myocyte, showing key proteins mutated in dilated cardiomyopathy (red labels), hypertrophic cardiomyopathy (blue labels), or both (green labels). May-2015-CSBRP 27
- 26. Causes and consequences of dilated and hypertrophic cardiomyopathy May-2015-CSBRP 28
- 27. DCM - MORPHOLOGY • Heavy (2-3x normal) and flabby heart • Mural thrombi – Thromboembolism • Valvular regurgitation Histologic abnormalities in DCM are nonspecific – Hypertrophy – Nucleomegaly, irregular nuclei – Endocardial fibrosis May-2015-CSBRP 29
- 29. What is “cor bovinum” ? • Bovine heart [>1000gm] • Seen in Syphilis, • AR due to other causes • Dilated cardiamyopathy May-2015-CSBRP 31
- 30. Name some causes for globular heart? • Wet Beri beri • AR due to any cause [Tertiary Syphilis] • Dilated cardimyopathy • Chaga’s disease May-2015-CSBRP 32
- 31. Arrhythmogenic right ventricular cardiomyopathy (ARVC) • Inherited disease, AD • Right ventricular failure and rhythm disturbances – Ventricular tachycardia or fibrillation • Sudden death Morphology: • Fatty infiltration and • Fibrosis May-2015-CSBRP 33
- 32. Arrhythmogenic right ventricular cardiomyopathy (ARVC) May-2015-CSBRP 34
- 33. Name some causes for fatty heart? [Fatty change of the heart] • Anemia • ARVH • Diphtheria May-2015-CSBRP 35
- 34. Hypertrophic Cardiomyopathy • Inherited disease, AD • Many genes are involved – HCM is a disease caused by mutations in proteins of the sarcomere – Most common is the gene encoding β-myosin heavy chain (β-MHC) • Myocardial hypertrophy • Poorly compliant LV > defective diastolic filling • Intermittent ventricular out flow obstrction May-2015-CSBRP 36
- 36. Restrictive Cardiomyopathy • Characterized by a primary decrease in ventricular compliance, resulting in impaired ventricular filling during diastole • Causes: – Radiation fibrosis – Amyloidosis – Sarcoidosis – Metastatic tumors – inborn errors of metabolism May-2015-CSBRP 38
- 37. Restrictive Cardiomyopathy Several other restrictive conditions: Endomyocardial fibrosis Loeffler endomyocarditis Endocardial fibroelastosis May-2015-CSBRP 39
- 39. E N D May-2015-CSBRP 41
Editor's Notes
- The minimal pathologic criteria for the diagnosis of systemic HHD are: Left ventricular hypertrophy (usually concentric) in the absence of other cardiovascular pathology and A clinical history or pathologic evidence of hypertension in other organs (e.g., kidney)
- Hypertension induces left ventricular pressure overload hypertrophy, initially without ventricular dilation. As a result, the left ventricular wall thickening increases the weight of the heart disproportionately to the increase in overall cardiac size (Fig. 12-20A). The thickness of the left ventricular wall may exceed 2.0 cm, and the heart weight may exceed 500 gm. In time the increased thickness of the left ventricular wall, often associated with increased interstitial connective tissue, imparts a stiffness that impairs diastolic filling, frequently with consequent left atrial enlargement.Microscopically, the earliest change of systemic HHD is an increase in the transverse diameter of myocytes, which may be difficult to appreciate on routine microscopy. At a more advanced stage variable degrees of cellular and nuclear enlargement become apparent, often accompanied by interstitial fibrosis.
- Thickening of the moderator band, the muscle bundle that connects the ventricular septum to the anterior right ventricular papillary muscle.
- Pulmonary Embolism ECG The most common ECG finding in the setting of a pulmonary embolism is sinus tachycardia, however the "S1Q3T3" pattern of acute cor pulmonale is classic. This is termed the McGinn-White sign. A large S wave in lead I, a Q wave in lead III, and an inverted T wave in lead III indicates acute right heart strain. This pattern only occurs in about 10% of people with pulmonary embolisms and is similar to the ECG findings in a left posterior fascicular block (LPFB). Recall that sinus tachycardia is actually the most common ECG finding during a pulmonary embolus.
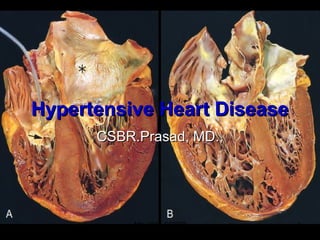
- Figure 12-20 Hypertensive heart disease, systemic and pulmonary. A, Systemic (left-sided) hypertensive heart disease. There is marked concentric thickening of the left ventricular wall causing reduction in lumen size. The left ventricle and left atrium (asterisk) are on the right in this apical four-chamber view of the heart. A pacemaker is present in the right ventricle (arrow). B, Pulmonary (right-sided) hypertensive heart disease (cor pulmonale). The right ventricle is markedly dilated and has a thickened free wall and hypertrophied trabeculae (apical four-chamber view of heart, right ventricle on left). The shape of the left ventricle (to the right) has been distorted by the enlarged right ventricle.
- “Cardiomyopathies are a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic. Cardiomyopathies either are confined to the heart or are part of generalized systemic disorders, often leading to cardiovascular death or progressive heart failure-related disability.”
- “Cardiomyopathies are a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic. Cardiomyopathies either are confined to the heart or are part of generalized systemic disorders, often leading to cardiovascular death or progressive heart failure-related disability.”
- genetic causes, including mutations in myocardial proteins involved in contraction, cell-cell contacts, and the cytoskeleton. These, in turn, lead to abnormal contraction or relaxation, or to dysregulated ion transport across cell membranes.
- ARVH = arrhythmogenic right ventricular (hypertrophy) cardiomyopathy
- Figure 12-29 The three major morphologic patterns of cardiomyopathy. Dilated cardiomyopathy leads primarily to systolic dysfunction, whereas restrictive and hypertrophic cardiomyopathies result in diastolic dysfunction. Note the changes in atrial and/or ventricular wall thickness. Ao, Aorta; LA, left atrium; LV, left ventricle.
- Genetic Influences. DCM is familial in at least 30% to 50% of cases, in which it is caused by mutations in a diverse group of more than 20 genes encoding proteins involved in the cytoskeleton, sarcolemma, and nuclear envelope (laminin A/C). In particular, mutations in TTN, a gene that encodes titin (so-called because it is the largest protein expressed in humans), may account for approximately 20% of all cases of DCM (Fig. 12-30). In the genetic forms of DCM, autosomal dominant inheritance is the predominant pattern; X-linked, autosomal recessive, and mitochondrial inheritance are less common. In some families there are deletions in mitochondrial genes that result in defects in oxidative phosphorylation; in others there are mutations in genes encoding enzymes involved in β-oxidation of fatty acids. Mitochondrial defects typically manifest in the pediatric population, while X-linked DCM typically presents after puberty and into early adulthood. Childbirth. A special form of DCM, termed peripartum cardiomyopathy, can occur late in pregnancy or up to months postpartum. The mechanism underlying this entity is poorly understood but is probably multifactorial. Pregnancy-associated hypertension, volume overload, nutritional deficiency, other metabolic derangements, or an as yet poorly characterized immunological reaction have been proposed as causes. Supraphysiologic stress can also result in DCM. This can happen with persistent tachycardia, hyperthyroidism, or even during development, as in the fetuses of insulindependent diabetic mothers. Excess catecholamines, in particular, may result in multifocal myocardial contraction band necrosis that can eventually progress to DCM. This can happen in individuals with pheochromocytomas, tumors that elaborate epinephrine (Chapter 24); use of cocaine or vasopressor agents such as dopamine can have similar consequences. Such “catecholamine effect” also occurs in the setting of intense autonomic stimulation, for example, secondary to intracranial lesions or emotional duress.
- Thus, takotsubo cardiomyopathy is an entity characterized by left ventricular contractile dysfunction following extreme psychological stress; affected myocardium may be stunned or show multifocal contraction band necrosis. For unclear reasons, the left ventricular apex is most often affected leading to “apical ballooning” that resembles a “takotsubo,” Japanese for “fishing pot for trapping octopus” (hence, the name)
- Thus, takotsubo cardiomyopathy is an entity characterized by left ventricular contractile dysfunction following extreme psychological stress; affected myocardium may be stunned or show multifocal contraction band necrosis. For unclear reasons, the left ventricular apex is most often affected leading to “apical ballooning” that resembles a “takotsubo,” Japanese for “fishing pot for trapping octopus” (hence, the name)
- Figure 12-30 Schematic of a myocyte, showing key proteins mutated in dilated cardiomyopathy (red labels), hypertrophic cardiomyopathy (blue labels), or both (green labels). Mutations in titin (the largest known human protein at approximately 30,000 amino acids) account for approximately 20% of all dilated cardiomyopathy. Titin spans the sarcomere and connects the Z and M bands thereby limiting the passive range of motion of the sarcomere as it is stretched. Titin also functions like a molecular spring, with domains that unfold when the protein is stretched and refold when the tension is removed, thereby impacting the passive elasticity of striated muscle
- Figure 12-31 Causes and consequences of dilated and hypertrophic cardiomyopathy. Some dilated cardiomyopathies and virtually all hypertrophic cardiomyopathies are genetic in origin. The genetic causes of dilated cardiomyopathy involve mutations in any of a wide range of genes. They encode proteins predominantly of the cytoskeleton, but also the sarcomere, mitochondria, and nuclear envelope. In contrast, all of the mutated genes that cause hypertrophic cardiomyopathy encode proteins of the sarcomere. Although these two forms of cardiomyopathy differ greatly in subcellular basis and morphologic phenotypes, they share a common set of clinical complications. LV, left ventricle.
- The average weight of the normal heart in man is about 300 gm. Through athletic exertion it can attain a weight of 500 gm. This increase is due to an extension of physiologic growth, with the fibers increasing in length and width but not increasing in number.
- Figure 12-32 Dilated cardiomyopathy. A, Four-chamber dilatation and hypertrophy are evident. There is a mural thrombus (arrow) at the apex of the left ventricle (on the right in this apical four-chamber view). The coronary arteries were patent. B, Histologic section demonstrating variable myocyte hypertrophy and interstitial fibrosis (collagen is highlighted as blue in this Masson trichrome stain).
- Figure 12-33 Arrhythmogenic right ventricular cardiomyopathy. A, Gross photograph, showing dilation of the right ventricle and near-transmural replacement of the right ventricular free-wall by fat and fibrosis. The left ventricle has a virtually normal configuration in this case, but can also be involved by the disease process. B, Histologic section of the right ventricular free wall, demonstrating replacement of myocardium (red) by fibrosis (blue, arrow) and fat (Masson trichrome stain).
- Figure 12-33 Arrhythmogenic right ventricular cardiomyopathy. A, Gross photograph, showing dilation of the right ventricle and near-transmural replacement of the right ventricular free-wall by fat and fibrosis. The left ventricle has a virtually normal configuration in this case, but can also be involved by the disease process. B, Histologic section of the right ventricular free wall, demonstrating replacement of myocardium (red) by fibrosis (blue, arrow) and fat (Masson trichrome stain).
- Figure 12-34 Hypertrophic cardiomyopathy with asymmetric septal hypertrophy. A, The septal muscle bulges into the left ventricular outflow tract, and the left atrium is enlarged. The anterior mitral leaflet has been reflected away from the septum to reveal a fibrous endocardial plaque (arrow) (see text). B, Histologic appearance demonstrating myocyte disarray, extreme hypertrophy, and exaggerated myocyte branching, as well as the characteristic interstitial fibrosis (collagen is blue in this Masson trichrome stain).
- Thus, takotsubo cardiomyopathy is an entity characterized by left ventricular contractile dysfunction following extreme psychological stress; affected myocardium may be stunned or show multifocal contraction band necrosis. For unclear reasons, the left ventricular apex is most often affected leading to “apical ballooning” that resembles a “takotsubo,” Japanese for “fishing pot for trapping octopus” (hence, the name)