This document discusses pulmonary drug delivery systems and recent developments. It begins by providing background on targeting drug delivery into the lungs and how this has become an important aspect of drug delivery systems. Various techniques for delivering drugs into the lungs are discussed, including inhalation therapy using metered dose inhalers and dry powder inhalers. The use of drug carriers like liposomes, nanoparticles, and microparticles to convey drugs to the lungs is also described. Recent developments in transitioning from CFC to HFA propellants for inhalers are summarized. The document concludes by outlining considerations for regional drug delivery to the lungs and respiratory tract anatomy.









![
. . .
2. Immunosuppressants
The immunosuppressant agent cyclosporine A (CsA) was effectively incorporated into
liposomes composed of egg yolk phosphatidylcholine (EYPC) with a molar ratio of 1:12
CsA/EYPC.31 The association percentage was high (95%).The generation of small aerosol
particles of CsA liposomes had no effect on CsA biological activity because CsA liposomes
were as effective as CsA resuspended in its normal carrier,Cremophor EL,in the inhibition
of anti-CD3 antibody stimulation of mouse spleen cell, as measured by the incorporation
of [3H] thymidine. CsA liposome particles have a mass median aerodynamic diameter of
2 µm, which permits distribution of the drug throughout the respiratory tract. Liposomes
containing CsA were given by aerosol for 15 minutes to mice, and the CsA concentration
in the lungs was found to be equivalent to that of a single daily i.v. injection 16 times more
concentrated (Fıg. 7). CsA liposomes can be produced and aerosolized in order to achieve
pulmonary concentrations with enough immunosuppressant activity to be effective in the
treatment of lung diseases.
Waldrep et al.32 proposed an optimum liposome formulation for nebulization contain-
ing glucocorticoids or immunosuppressant, using dilauroylphosphatidylcholine (DLPC)
alone instead of dipalmitoylphosphatidylcholine (DPPC), dimyristoylphosphatidylcholine
(DMPC), or egg yolk phosphatidylcholine (EYPC).
Liposomes of DLPC containing concentrated amounts of CsA and budesonide (Bud)
FIGURE 7. Comparison of CsA concentrations in blood and lung tissue after 4 days of small-particle
aerosol or intravenous administration of CsA-containing liposomes. Liposomes were composed of 2
mg of CsA/ml and 15 mg of phosphatidylcholine/mL. Three mice (26 g) were used at each time point.
Drug was administered by aerosol for 2 h twice daily, giving a dose of 1.8 mg of CsA/kg ([25 µg of
CsA/L of aerosol × 0.026 L/min {min vol} × 240 min × 0.3 {retention factor}]/0.026 kg), or for 15 min
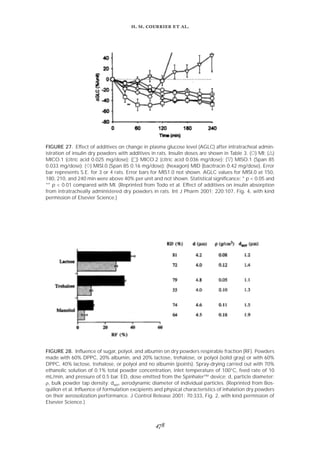
once daily, giving a dose of 0.11 mg of CsA/kg. Intravenous administration was a single daily injection
of 0.1 mL of CsA liposomes in the tail vein, giving a dose of 1.8 mg of CsA/kg. CsA tissue concentra-
tions were determined by HPLC. (Reprinted from Gilbert et al. Characterization and administration of
cyclosporine liposomes as a small-particle aerosol. Transplantation 1993; 56(4):976, Fig. 1, with kind
permission from Lippincott Williams & Wilkins.)](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-14-320.jpg)
![
have been formulated and nebulized.33 Formulations 40 times more concentrated than com-
mercial ones and used by nebulization of Bud suspensions could both reduce nebulization
time and improve patient compliance.The optimum DLPC/CsA and DLPC/Bud propor-
tions are 1:7.5 and 1:15, respectively. With these, liposomes of 1–3 µm diameter could be
formulated, and after nebulization their sizes were reduced (270–560 nm).
After the inhalation of DLPC/CsA nebulized liposomes, their biodistribution was
studied in mice.34 In this study,on a per-gram-tissue basis,the lung contained approximately
18 times more CsA than the liver, and 104 times more CsA than the blood, demonstrating
the effective pulmonary targeting of the CsA/DLPC liposome aerosol.The in vitro immu-
nosuppressant effect of CsA isolated from pulmonary tissue,following delivery of nebulized
DLPC/CsA liposomes, was maintained. Inhibition (99%) of [3H]TdR by antigen-specific
stimulation reduction was revealed, along with inhibition (95%) of mitogen sensitivity.This
DLPC/CsA formulation is promising and could be used to treat chronic asthma and al-
lergies.
Liposome vectors and CsA dissociation were studied in mice following pulmonary
delivery.35 A stable radioactive complex of 99mTc-liposomes DLPC/CsA was delivered by
intratracheal (i.t.) instillation. The 99mTc-liposomes DLPC vector was retained 17 times
longer than the half-life of CsA in a normal lung and 7.5 times longer than in an inflamed
lung (Table 1).
Studies on dogs were carried out, selectively observing the immunosuppressant effect
on the lung of the aerosolized form of CsA, with the aim of seeing whether this system is
suitable for pulmonary transplants,which are compromised by chronic and acute rejection.36
The lungs absorb the nebulized CsA liposomes faster than the other organs do with weaker
concentrations of CsA. In this model, the retention of the CsA delivered by the liposomes
in the lungs was around 120 minutes.
3. Glucocorticoids
Liposomes composed of 1,2-distearoyl phosphatidylcholine (DSPC) and 1,2-distearoyl
phosphatidylglycerol (DSPG) were prepared in order to incorporate triamcinolone aceton-
TABLE 1. Half-Lives in Normal and Inflamed Lungsa
Components T1/2 α
CsA - normal lungs 17.0 ± 3.8 min
CsA - inflamed lungs 17.6 ± 7.3 min
liposomes DLPC - normal lungs 4.8 ± 0.1 h
liposomes DLPC - inflamed lungs 2.2 ± 0.9 h
HSA - normal lungs 4.2 ± 2.4 h
HSA - inflamed lungs 2.0 ± 0.3 h
a 99mTc-cyclosporine A (CsA), 99mTc-liposomes composed of DPPC, and 99mTc-
human serum albumin (HSA) (Arppe et al., 1998).](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-15-320.jpg)
![
. . .
ide phosphate (TAP).37 The glucocorticoid was in its hydrophilic form so that the liposome
membrane acts as a barrier and permits slow delivery.A liposome incorporating a lipophilic
glucocorticoid quickly slackens under unbalanced conditions (dilution,administration).These
liposomes are stable for 24 hours in contact with physiological fluid.Seventy-five percent of
TAP remains encapsulated, the initial encapsulation rate being 7–8.5%. Administration of
TAP solution and TAP-liposomes (207 ± 16 nm) i.t. and i.v. was compared in rats.The i.t.
administration ofTAP-liposomes enables prolonged occupation of glucocorticoid receptors,
compared with i.v. administration or with treatment with a TAP solution. Its cumulative
effect was 1.6 times higher in the lungs than in the liver.
Liposomes of EYPC–cholesterol (CHOL) incorporating dexamethasone palmitate
(DEXP),in a molar proportion of 4:3:0.3,were studied.38 Encapsulation of the DEXP was
effective (70%) in comparison with its nonesterified form (<2%).The biological activity of
DEXP was evaluated on blood mononuclear cells over a 24-hour period,measuring its anti-
lymphocyte proliferation properties and its inhibition of interferon-γ production (Table 2).
The DEXP incorporated in the liposomes kept its biological activity. Nebulization studies
in animals should confirm whether this vector is promising in drug delivery to the lungs.
DPPC liposomes containing dexamethasone (DEX) in a molar proportion of 9:1
were prepared and instilled by the i.t. route in rats.39 Encapsulation was effective (35%),
and the size of the liposome-entrapped dexamethasone (L-DEX) was approximately 231
± 32 nm. The pulmonary and blood retention levels of [3H]DEX radioactive compound
were, respectively, 50% and 1% for L-DEX and 26% and 5% for the free DEX 1.5 hours
after instillation. Its effects on reduction of white blood cell levels in peripheral blood and
of adrenocorticotropic hormone (ACTH) levels in the plasma were studied. L-DEX has a
prolonged action (>72 h) on reduction of white blood cells,whereas free DEX has no more
effect after 24 hours.Plasma ACTH levels are less significantly reduced with L-DEX (60%
in 1 h, 25% in 72 h) than with free DEX (80% in 1 h, 50% in 72 h). This study showed
that the retention of dexamethasone delivered directly into the lungs in liposomal form
was significantly prolonged (prolonged anti-inflammatory action) and that the side effects
were reduced.
Following these encouraging results, Suntres et al.40 examined the prophylactic effect
of L-DEX in an animal pulmonary model damaged by lipopolysaccharides (LPS).40 The
LPS stimulate the phagocytes to generate metabolites, which play a significant role in lung
pathogenesis.Rats were pre-treated by the i.t.route with L-DEX,DEX,or a saline solution,
then treated by the i.v.route with LPS.Measurements of the activity of various markers were
taken in: pulmonary cells (endothelial capillary cell markers,such as angiotensin-converting
TABLE 2. Inhibition (%) of Concavalin A Stimulating Proliferation of Lymphocytes and
Production of Interferon γ ( INF-γ )a
Inhibition %
concavaline A-stimulating Free DEXP Liposome-DEXP
Lymphocytes proliferation 94 94
INF-γ production 96 96
a Induced by 10–6 M of free dexamethasone palmitate (DEXP) or by DEXP loaded liposomes composed of EPC-
Cholesterol. (Benameur et al., 1995.)](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-16-320.jpg)
![
enzyme [ACE] and type-II alveolar epithelial cell markers, such as alkaline phosphatase
[AKP]), inflammatory response markers (myeloperoxidase [MPO] and elastase activity,
chloramine concentration) and pro-inflammatory mediators (concentration of A2 phospholi-
pase,leukotriene eicosanoid B4,and thromboxane B2 in plasma and histamine in the lungs).
L-DEX was more effective than DEX and protected the pulmonary cells from the LPS.The
ACE and AKP activities were reduced by only 5% and 18%,respectively,while DEX reduces
them by 20 and 28%, respectively. DEX inhibited the increase in inflammatory mediator
activities. L-DEX was 15% more effective in the reduction of MPO (55%) and elastase
(68%) than DEX and 20% more effective in the reduction of chloramine (50%).The three
pro-inflammatory mediators studied are also inhibited by L-DEX and DEX: phospholipase
A2 (62 vs. 45%), leukotriene eicosanoid B4 (76 vs. 64%), and thromboxane B2 (76 vs. 64%)
in plasma.Suntres et al.40 also highlighted that pretreatment with saline solutions and blank
liposomes does not inhibit the effects induced by treatment with LPS.
4. Corticosteroids
The tolerance and safety of DLPC liposomal aerosols containing beclomethasone dipropio-
nate (BDP) were studied in 10 healthy volunteers.34 According to pulmonary function and
blood tests, exposure to aerosols containing amounts of BDP equivalent to or double those
managed by metered dose inhaler (MDI) and dry powder inhaler (DPI) for the treatment
of asthma was well tolerated.
The pulmonary distribution and clearance of DLPC-BDP liposomes and DPPC-BDP
liposomes were compared in 11 healthy volunteers.41 DLPC formed liposomes suitable for
atomization.33 Because DPPC is the major component of pulmonary surfactant and is used
for respiratory distress syndrome (RDS) therapy,9 this should also be investigated. DLPC
and DPPC liposomes had sizes of 3.5 µm and 5.0 µm,respectively,before atomizing and 0.8
µm and 0.9 µm, respectively, after atomizing.The total outputs of the nebulized liposomes
were 11.4 µg with DLPC liposomes and 3.1 µg with DPPC liposomes. This difference
could be due to phase transition temperatures (DLPC –2°C, DPPC +41°C). DPPC could
produce more rigid liposomes, which would find it difficult to pass through the openings of
the atomizer. Clearance of 99mTc-liposomes complexes was relatively slow: 24 hours after
inhalation,79% of the radioactivity originally deposited was detected using DLPC and 83%
using DPPC. Both formulations were suitable for the encapsulation of drugs because they
offered a delivery tolerated by the lower respiratory tracts. However, atomization was more
effective with the DLPC liposomes.
Liposomes containing BDP were prepared in different manners in order to improve
their stability.42 After preparation,the liposomes were freeze-dried and then rehydrated just
before atomizing. Of the series of lipids (DLPC, DMPC, DPPC, HSPC), DLPC, used
previously, was shown to be the most effective for the encapsulation of BDP, although the
encapsulation rate remains low (MLVs: 3.69 ± 0.10% m/m and SMLVs: 2.03 ± 0.08%).
Despite being increased in size after freeze-drying and rehydration, DLPC liposomes were
the smallest liposomes produced: 10.30 ± 1.35 µm and 3.87 ± 0.20 µm, before and after
atomizing,respectively.Atomization made it possible to reduce their size by breaking up any
aggregates.The best atomizing output is obtained with DLPC (78.3%),whereas the DPPC
liposomes have incorporated 25% of BDP.The RF of the DLPC liposomes was 75%,which
was 10% higher than that of the other lipids.](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-17-320.jpg)


![
. . .
mulation was achieved when the hydrophobic interactions between the acryl chain of the
lipid and INF-γ were the strongest. Hydrogenated soybean phosphatidylcholine (HSPC),
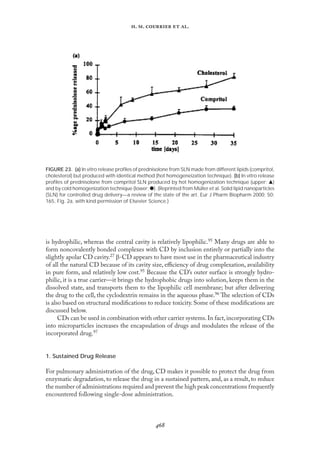
distearoyl--α-phosphatidylcholine (DSPC), and distearoyl--α-phosphatidylglycerol
(DSPG) provided stability in the following formulation: HSPC/DSPG 10:1 and DSPC/
DPPG 10:1. Fınally, INF-γ can be nebulized thanks to the liposomes, which absorb INF-γ
on their surface (Table 3).
The prophylactic effect of an INF-γ and synthetic double-stranded polyriboinosinic-
polyribocytidylic acid (poly IC) stabilized with poly--lysine:carboxymethylcellulose (LC)
(poly[ICLC]) encapsulated in a liposomal formulation was highlighted in mice infected by
a lethal amount (10 LD50) of the influenza virus.50 The immunomodulator-liposomes were
administered intranasally, but direct lung administration is feasible.
c. Interleukin
Human serum albumin and interleukin 2 (IL-2)–loaded DMPC liposomes, as well as
free IL-2, were nebulized in dogs51 in order to compare the immunological activation
of various IL-2 formulations. A toxicity assessment revealed no side effects for either
treatment. The bronchoalveolar lavage (BAL) leukocyte cell count increased significantly
after inhalation of IL-2–liposomes versus inhalation of free IL-2. A greater proportion
of lymphocytes and eosinophils was observed after IL-2–liposomes treatment. Nontoxic
activation of pulmonary immune effectors for treating cancer in the lung may be possible
using IL-2–liposomes.
DMPC liposomes containing IL-2 were administered by aerosol in several immuno-
deficient patients.52 The rate of encapsulation,or at least of association,was very high (98.8%),
and the average diameter of these liposomes was around 1.1 µm. Patient compliance, safety,
toxicity,and the immune effects of IL-2 liposomes were studied in individuals with primary
immune deficiency and,subsequently,a larger cohort of patients with hepatitis C.According
to the authors of this study,a biological activity of aerosol IL-2 liposomes has been observed
in viral disease (hepatitis C), and additional studies on aerosol Il-2 liposomes in individuals
with hepatitis C and HIV are planned.
TABLE 3. Liposome Formulations Having Adsorbed INF-γ at Their Surface, To Have
Efficient Nebulization of INF-γa
Liposome
composition
Size of liposomes
(nm, average ± SD)
Size of aerosols
(µm, average ± SD)
% of recovery
remaining
% of recovery
aerosolized
None 3.06 ± 1.99 3.1 ± 0.7 0.4 ± 0.2
HSPC/DSPG (10/1) 45.0 ± 24 4.88 ± 2.84 27.2 ± 4.7 25.7 ± 12.6
DSPC/DPPG (10/1) 28.5 ± 19 — 29.8 ± 2.6 43.1 ± 16.6
EPC/DSPG (10/1) 43.7 ± 23 3.79 ± 2.29 16.2 ± 13.0 15.8 ± 2.6
EPC 40.8 ± 24 4.99 ±3.06 3.7± 1.0 1.2 ± 0.4
a Kanaoka et al., 1999](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-20-320.jpg)


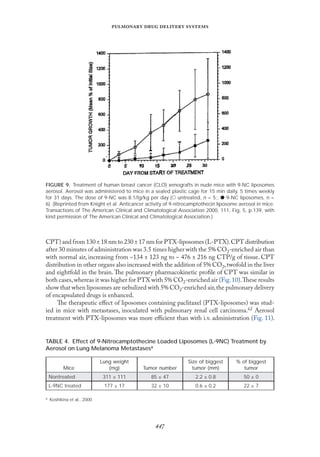
![
The weight of the lungs and the number of visible tumors decreased by ~26% and ~ 32%,
respectively, compared with the untreated mice.Their life expectancy also increased, by ~10
days. This study reveals the potential therapeutic application of aerosols for the treatment
of pulmonary cancer.
11. Bioadhesive Liposomes
Bioadhesive drug delivery systems were introduced in order to prolong and intensify the con-
tact between controlled delivery forms and the mucous apical pole,inducing active transport
processes.63 Contact with the mucus of the epithelium is called muco-adhesion, and direct
contact with the cellular membrane is called cyto-adhesion. Lectins are nonimmunological
glycoproteins that have the capacity to recognize and bind to glycoproteins exposed at the
epithelial cell surface.
Liposomes functionalized with lecithins appeared to be capable of improving their bind-
ing to human alveolar cells (A549 and primary cells).64 In this study, the unfunctionalized
liposome formulation was optimized by measuring the loss of carboxyfluorescein (CF) loaded
in the liposomes during atomization.Liposomes composed of DPPC–CHOL (50–50% mol)
were more stable during atomization (8% CF loss) than DPPC liposomes (15–20% CF loss),
even in the presence of pulmonary surfactant. Lehr et al.63 reported that the atomization
of DPPC–CHOL liposomes with lecithin functionalization did not significantly influence
their physical stability.The cell-binding capacity of functionalized liposomes is much higher
than that of unfunctionalized liposomes, even after atomization (Fıg. 12).
Immunoliposomes—liposomes carrying specific antibodies—can target cells carrying a
specific antigen. Margalit65 reported that they have been used to target pulmonary tumors
in vitro and in vivo.
12. Dry Powder Liposomes
An optimum formulation of dehydrated liposomes depends on several factors: the liposome
composition, the presence of cholesterol (CHOL), the incorporation of a cryoprotective
sugar, the preparation method, and the nature and proportion of the incorporated drug. An
optimum liposome formulation corresponds to an optimum size, lamellarity (unilamellar
[UL] or multilamellar [ML]), has a maximum drug incorporation efficiency and oxidation
index. An optimum dry powder formulation is characterized by its repose angle, its com-
pressibility index, and its dispersible and respirable fractions.
In the past, several formulations of liposome dry powder inhaler (DPI) have been de-
veloped. Among these, a formulation of liposome DPI containing anti-asthmatic ketotifen
fumarate (KF),was optimized.25 Liposomes formed by two successive hydrations before and
after sonification (1 and 2 hours, respectively) and with a molar composition KF/(EYPC-
CHOL) (1:15) demonstrate a maximum encapsulation rate.In this case,sucrose is revealed
to be the best system cryoprotector, with a mass lipid/sugar ratio of 1:12 and a maximum
concentration of 500 mM. When lactose monohydrate (Sorbolac-400) was added before
freeze-drying, 97.92 ± 0.54% KF retention was achieved. The oxidation of liposome lipids
is not inhibited by the presence of nitrogen or antioxidant agents, with the oxidation index
increasing from 0.427 ± 0.01 to 1.510 ± 0.01 (Table 5).Fınally,the respirable fraction of this](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-25-320.jpg)
![
. . .
formulation (21.59 ± 1.53%) was comparable with a commercial control (26.49 ± 1.52%).
The KF-liposome DPI was successfully prepared according to the respirable fraction to be
delivered to the central and peripheral pulmonary tract. Obviously, the choice of the cryo-
protector is dependent on the chemical structure of the drug. For example, as a reducing
sugar, the sucrose would be entirely unsuitable for protein or peptide delivery.
Table 5. Formulation of Dry Powder Inhaler (DPI) Liposomesa
Formulations KF : EYPC : CHOL Size (µm)
% of
encapsulated KF Oxidation index
KF[1] 1 : 15 : 0 1.56 ± 0.26 86 1.510 ± 0.01
KF[2] 1 : 10 : 5 1.70± 0.12 70 1.425 ± 0.01
KF[3] 1 : 7.5 : 7.5 2.05 ± 0.10 64 1.328 ± 0.01
a Liposomes are composed of egg yolk phosphatidylcholine (EYPC) and cholesterol (CHOL), which permit the
highest ketotifen fumarate (KF) incorporation rate, with an oxidation index that is still high (Joshi and Misra25).
FIGURE 12. Interaction of lectin-functionalized liposomes with alveolar epithelial cells. Cell association
of 200 µg wheat germ agglutinin (WGA)-liposomes with A549 cells. WGA liposomes = WGA-functional-
ized liposomes; blank liposomes = DPPC:cholesterol liposomes; WGA liposomes + free WGA = WGA
liposomes and 20-fold free WGA; inhibitory sugar = 20 µl of 20.0 mM diacetylchitobiose; LS = alveoafact
(lung surfactant). Results represent the average and standard deviation of at least 3 determinations
from 2 different passage numbers for A549 cells. (Reprinted from Abu-Dahab et al. Lectin-functional-
ized liposomes for pulmonary drug delivery: effect of nebulization on stability and bioadhesion. Eur J
Pharm Sci 2001; 14:43, Fig. 6b, with kind permission of Elsevier Science.)](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-26-320.jpg)

![
. . .
rate fell as the albumin ratio increased. In conclusion, sustained-release microspheres were
suitable for dry powder inhaled pulmonary drug delivery systems.
b. Target or Avoid Alveolar Macrophages?
Targeting drugs to alveolar macrophages has the distinct advantage of delivering high concen-
trations of drug to a cell that plays a central role in the progression of disease (tuberculosis)
and in immune responses.
The microspheres can target alveolar macrophages (AMs) without eliciting a pulmonary
inflammatory response in vitro.22 In fact, a cell culture of AM, in the presence of micro-
spheres composed of PLA, produces negligible quantities of oxidants and tumor necrosis
factor alpha (TNF-α) inflammatory cytokines. Interactions between PLA microspheres,
marked by rhodamine 6G,which is a fluorescent agent,and AM are concentration-dependent
(~30% interactions with a concentration of 50,000 particles /mL). Endocytosis of the mi-
crospheres was revealed in the presence of certain endocytosis inhibitors—lysosomotropic
agents, NH4Cl, and chloroquine—reducing AM–particle interaction by around 50%. This
study demonstrated that microspheres can enter alveolar macrophages without activating
them, thus enabling possible drug delivery to target macrophages, for example, in the case
of tuberculosis.
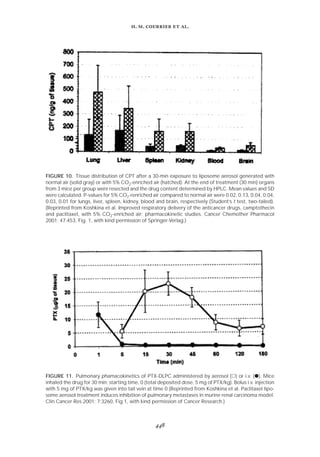
Wang et al.71 showed that the coencapsulation of an immunomodulator (monophos-
phoryl lipid A [MPLA]) in PLGA microspheres makes it possible to increase the rate of
phagocytosis (Fıg. 13). In the case of other diseases, alveolar macrophages must be avoided
FIGURE 13. Effect of coencapsulated MPLA on phagocytosis of PLGA microspheres containing plasmid
DNA. J774A-1 cells were incubated with PLGA microspheres (6000 g/mole) containing MPLA (᭜) or
no MPLA () for 0.75, 1.5, 3, 6, 12 and 24 h. Free microspheres were removed by PBS washing, cells
were fixed, and the number of microspheres per cell was counted by phase contrast microscopy. Error
bars indicate S.D. (n = 3). (Reprinted from Wang et al. Encapsulation of plasmid DNA in biodegradable
poly(D,L-lactic-co-glycolic acid) microspheres as a novel approach for immunogene delivery. J Control
Release 1999; 57:16, Fig. 9b, with kind permission of Elsevier Science.)](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-28-320.jpg)
![
in order to prevent phagocytosis clearance and thus to enhance the alveolar half-life and
bioavailability of the drug.
DPPC plays a role in alveolar macrophage phagocytotic of microparticles.21 The in-
teractions of PLGA and DPPC/PLGA microspheres containing peroxidase, as a protein
model, have been evaluated on an AM cell culture by confocal microscopy. After incuba-
tion for 1 hour, the PLGA particles are located in the macrophage cytoplasm (95 ± 1.35%),
while the DPPC–PGLA particles are instead located at their surface (26.2 ± 13.9%). X-ray
photoelectron spectroscopy (XPS) results indicated that the inclusion of DPPC in the
microspheres altered the microsphere surface chemistry, with the DPPC covering a large
portion of the microsphere surface, but did not entirely mask the PLGA. The dominance
of DPPC on the microsphere surface was highly beneficial in moderating the interactions
occurring between the microspheres and phagocytic cells in the lung. DPPC reduced the
adsorption of opsonic proteins,thereby reducing microsphere phagocytosis occurring in the
alveoli, which enabled possible alveolar drug delivery (Table 6). These microspheres could
be designed to act as a controlled delivery system for small molecules, peptides, or proteins
for pulmonary administration.
Other studies were investigated to understand the inhibition of pulmonary phagocyto-
sis. In fact, respirable PGLA microspheres (2–3 µm) containing a fluorophore (rhodamine
B [RB]) were used as a model.20 RB’s loading efficiency was approximately 18%, and its
burst effect was very low, with less than 0.5% being released up to 19 hours. Two alveolar
macrophage types were used for this study: the NR8383 cell line and alveolar macrophages
(AM) freshly isolated from the lungs of rats. Seventy percent of the NR8383 population
phagocytosed a mean of 3.24 ± 0.69 microspheres per cell. The use of inhibitors (cytocha-
lasin D, Na azide) prevented phagocytosis. The phagocytosis of microspheres coated with
polaxomer 338 depended on the microspheres-per-cell ratio R. Compared to the control,
when R = 5, the phagocytosis reduction was 20% and 15% for AM and NR8383, respec-
tively; and when R ≥ 10,phagocytosis was 10–15% reduced for AM,while no reduction was
found for NR8383.The phagocytosis of microspheres coated with DPPC was significantly
lower than the control at all microsphere-per-cell ratios. Even at excess ratios, around 65%
of phagocytosis was inhibited for both cell types.
c. Importance of Encapsulated Drug Nature
El-Baseir et al.67 studied the in vitro release kinetics of nedocromil sodium (NS) (hydro-
soluble compound) and beclomethasone dipropionate (BDP) (hydrophobic compound) from
poly(-lactic acid) (PLA) microspheres. The release kinetics of NS exhibited a biphasic
TABLE 6. Effect of DPPC on Microparticle Internalization by Alveolar Macrophages
(AM)a
Particles PGLA PGLA/DPPC
Size (µm) 3.5 ± 1.72 3.3 ± 1.00
(%) of internalization in AM 65.1 ± 15.8 26.2 ± 13.9
a Evora et al.21](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-29-320.jpg)


![
f. Proteins
Calcitonin. The pulmonary administration and in vitro degradation of gelatin microspheres
loaded with salmon calcitonin (SC) was studied by Morimoto et al.74 Gelatin microspheres
made it possible to prevent particle degradation by enzymes. The in vitro release study
(Fıg. 16) revealed that SC seems to be dependent on the gelatin microsphere load and
not on the particle size. Within 2 hours, approximately 85% of SC was released from
positively-charged gelatin microspheres, while 40% was released from negatively-charged
gelatin microspheres. These results suggested that the SC released from the microspheres
depended on the electrostatic repulsion between SC (isoelectric point [IEP] = 8.3) and
positively charged gelatin microspheres (IEP = 9). However, the initial release of SC from
negatively-charged microspheres was suppressed by the formation of a poly–ion complex.
Consequently, the electrostatic forces relationship between the incorporated proteins and
gelatin may be an important factor affecting the release rate of incorporated proteins from
gelatin microspheres.
The results for intratracheal administration of SC-loaded gelatin microspheres are
given in Fıgure 17. The hypocalcemic effect following the administration of SC in both
types of gelatin microspheres was significantly greater than that following administration
in aqueous solution (in pH 7.0 PBS).The hypocalcemic effect following the administration
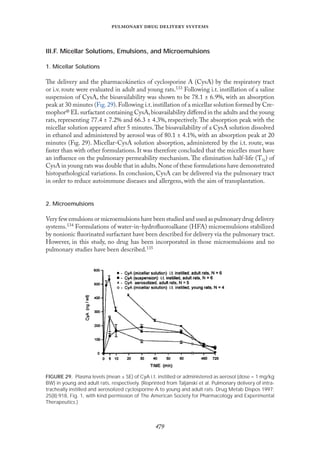
FIGURE 16. Release profiles of salmon calcitonin from gelatin microspheres with different charge
(A) and different particle sizes (B) in pH 7.0 PBS at 37°C. Positively charged microspheres: 11.2 µm
(᭝). Negatively charged microspheres: 10.9 µm (᭡). Each point represents the mean ± s.e.m., n = 4.
(Reprinted from Morimoto et al. Gelatin microspheres as a pulmonary delivery system: evaluation
of salmon calcitonin absorption. J Pharm Pharmacol 2000; 52:614, Fig. 2, with kind permission of
Pharmaceutical Press.)](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-33-320.jpg)




![
III.C. Solid Lipid Nanoparticles
Solid lipid nanoparticles (SLNs),introduced in 1991,represent an alternative carrier system
to traditional colloidal carriers, such as emulsions, liposomes, and polymeric micro- and
nanoparticles.94 Indeed,SLNs combine the advantages of the safety of lipids (lipids are well
tolerated by the body) and the possibility of large-scale production. Many different drugs
have been incorporated in SLN (Prednisolone, Diazepam, Camptothecin, etc.).The factors
determining the loading capacity of a drug in the lipids are the solubility of the drug in melted
lipid, the miscibility of drug melt and lipid melt, the chemical and physical structure of the
solid lipid matrix, and the polymorphic state of the lipid material. The drug incorporation
model may vary according to the preparation method. There are three drug incorporation
models (Fıg. 22), just as for polymeric microspheres and nanospheres: the solid solution
model (drug molecularily dispersed), the core-shell models with drug-enriched shell (lipid
core), and drug-enriched core (lipid shell).
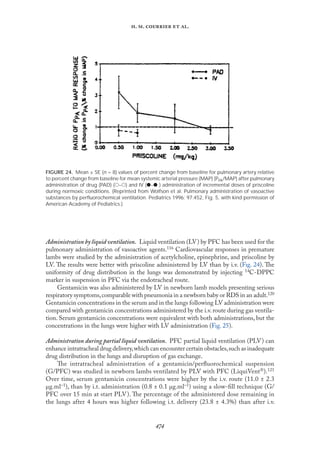
Controlled release of drugs and pulmonary administration. It is possible to modify release
profiles as a function of lipid matrix, surfactant concentration, and production parameters.
In vitro drug release could be achieved for up to 5–7 weeks.The profiles could be modulated
to demonstrate prolonged release without any burst at all, but also to generate systems with
different percentages of burst followed by prolonged release (Fıg.23).The release profiles are
not, or only slightly, affected by particle size. Because the release profile can be modulated,
controlled delivery of drug after pulmonary administration can be performed.For pulmonary
administration, SLN dispersions can be nebulized (without any significant change in mean
particle size), and SLN powders could be used in a DPI.
III.D. Cyclodextrins
Cyclodextrins (CDs) are cyclic nonreducing oligosaccharides containing 6, 7, or 8 gluco-
pyranose units (α-,β-,or γ-CD,respectively).The CD exterior,containing hydroxyl groups,
FIGURE 22. Three drug incorporation models (solid solution model (left), core-shell models with
drug-enriched shell (middle), and drug-enriched core (right). (Reprinted from Müller et al. Solid lipid
nanoparticles [SLN] for controlled drug delivery—a review of the state of the art. Eur J Pharm Biopharm
2000; 50:167, Fig. 5, with kind permission of Elsevier Science.)](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-43-320.jpg)





![
dry powders appear to present bioactivity between 40 and 66% and bioavailability of around
29% compared to s.c. injections. The pulmonary delivery of peptides and proteins by dry
powder should be a future therapy.
An alternative micronization technique using an aerosol solvent extraction system
(ASES) has been studied to avoid the insufficient brittleness of crystals that can occur
when using normal micronization.130 Several steroids were dissolved in an organic solvent
(CH2Cl2 or MeOH) and sprayed into supercritical carbon dioxide. Crystallinity studies
were then carried out. Budenoside and triamcinolone acetonide (TCA) demonstrated no
change in crystallinity, with or without the addition of surface-active phosphatidylcholine
(PC).While the addition of PC to prednisolone led to an amorphous powder,PC tended to
decrease particle size but to increase wettability. Residual solvent containing microparticles
was found to be less than 350 ppm in all cases. A median particle-size diameter was found
to be less than 5 µm and thus within the respirable range.
5. Solutions and Dry Powder Additives
Drugs composed of dry powders or solutions can be managed in a more effective way when
combined with additives.The pulmonary absorption of peptides or proteins from dry powder
or solution can be enhanced by using an additive.101 Salmon calcitonin (SC) was insufflated
in the form of solution or powder containing an additive,such as oleic acid,lecithin,or citric
acid.The absorption effect depends,first,on the additive concentration.With a dry powder,
the bioactivity of SC was around 34 ± 7%. By adding oleic acid, it increased to 58 ± 10%.
Conversely, additives in solution had almost no effect. Indeed, on an identical volume of
epithelial fluid,additives in the liquid form were less concentrated than in the powder form.
That is why oleic acid is more effective in powder form than in solution.This additive should
increase the paracellular permeability of the small junctions and enable the absorption of
peptides and proteins.
The effects of several additives were studied by i.t. administration of insulin in solution
and dry powder.131 Bacitracin and Span 85 are effective in supporting the hypoglycemic ef-
fect induced following the administration of insulin solutions.The effect lasted 180 minutes
after administration,and the insulin bioavailability was 100% compared to i.v.administration.
The citric acid supported the hypoglycemic effect induced following the administration of a
dry powder of insulin (Fıg.27).The effect lasted a longer period of 240 minutes,but a lower
insulin bioavailability was obtained—between 42 and 53%, depending on acid concentra-
tion. The insulin bioavailability was higher with citric acid than without (12%). Moreover,
the insulin powder containing citric acid was not toxic for the pulmonary cells. Citric acid
appears to be a potential additive for insulin powder absorption.
Studies to optimize the respirable fraction of particles inhaled by aerosol have been
conducted by determining the effects of the formulation and the physical characteristics of the
dry powder.132 When formulated with an appropriate composition (albumin/lactose/DPPC
[30:10:60 in weight]) and adequate physical characteristics,the powders exhibited excellent
aerosolization properties in the Andersen cascade impactor,with emitted doses reaching 90%
and respirable fractions up to 50% using the Spinhaler device, a first-generation inhaler
(Fıg. 28). The addition of albumin slightly increased the particle size (3–5 µm) and made
them more porous and less dense, and therefore easier to breathe in. These powders can
incorporate drugs such as peptides, proteins, or DNA for local and systemic delivery.](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-53-320.jpg)
![
. . .
Reverse water-in-chlorofluorocarbon micelles stabilized by lecithin and containing
peptides have been aerosolized.136 The surfactant concentration of the metered dose inhaler
(MDI) formulation should range between 0.5% and 2% (w/v), with the remaining volume
component being the propellant.Although this system was stable and able to deliver peptides
and proteins to the respiratory tract, its use should be limited because of the international
agreements following the Montreal protocol (1987) and calling for the total phase-out of
CFC production. Therefore, ozone-friendly propellants such as HFA, hydrocarbons, or
fluorocarbons should be used in MDI applications.
Reverse microemulsions stabilized by lecithin and using propane and dimethyl ether
as propellant gases have been described.137 These microemulsions, stable for more than 4
weeks at ambient temperature, had an aqueous internal phase of around 3 ± 2 µm diameter
and 36% respirable fraction.This report is the first to use lecithin reverse microemulsions for
pulmonary delivery of polar drugs.The use of reverse microemulsion (versus micelles) should
allow the solubilization of a greater quantity and variety of polar compounds. Extensive
characterization of aerosols generated by MDIs containing microemulsion is underway.
Reverse water-in-fluorocarbon emulsions stabilized by a semifluorinated amphophile
derived from dimorpholinophosphate CnF2n+1(CH2)mOP(O)[N(CH2CH2)2O]2 (FnHm-
DMP) made it possible to prepare stable water-in-fluorocarbon emulsions.138 The external
phase of these emulsions consisted of perfluorooctyl bromide (PFOB, perflubron), whereas
their internal phase contained the drugs solubilized or dispersed in water.These emulsions
are being investigated as pulmonary drug delivery systems,either for systemic or local deliv-
ery of drugs.139 Physicochemical studies have made it possible to select FnHmDMP as the
candidate yielding the most stable emulsions.140 Studies on the evaluation of FnHmDMP
and FnHmDMP-stabilized emulsion cytotoxicity have been investigated in vitro on mouse
fibroblasts and human lung epithelial cells.141 F8H11DMP and F10H11DMP were found
to be the most biocompatible semifluorinated surfactants (viability average: 88 ± 4% and >
100%,respectively at 1% w/v).In addition,some water-in-fluorocarbon emulsions stabilized
with F8H11DMP and F10H11DMP surfactants appear to be biocompatible for pulmonary
drug delivery (Fıg.30).Currently,the acute toxicity of water-in-PFOB emulsions,stabilized
by F8H11DMP, is being investigated in mice, as well as the delivery of insulin contained in
these emulsions administered by the i.t. route.
IV. TRANSITION TO CFC-FREE INHALERS
A. Aerosol generators
1. Technical Transition to CFC-Free Inhalers
Aerosol generators make it possible to administer a predetermined amount of drug into the
lungs. In order to specifically target the drugs, these devices have been extensively studied
and technically improved over the last decade and are described in the literature.142 They
include aerosol generators of (i) drug powders (Spinhaler, Cyclohaler, Turbuhaler); (ii)
autoactivated aerosols (Maxair, Prolair, Autohaler); (iii) spray diffusers (Pulmicort
Nebulization, Bricanyl).143](https://image.slidesharecdn.com/58291aeb-f00d-4c5b-b04b-8193ea8cabeb-150602205938-lva1-app6892/85/Crit-review2002-56-320.jpg)
















![Clinical Pharmacokinetics-I [half life, order of kinetics, steady state]](https://cdn.slidesharecdn.com/ss_thumbnails/clinicalpk-ihalflifeorderofkineticssteadystate-140217020044-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)








































![Pulmonary drug delivery system [PDDS]](https://cdn.slidesharecdn.com/ss_thumbnails/pulmonarydrugdeliverysystem-160619063517-thumbnail.jpg?width=640&height=640&fit=bounds)










