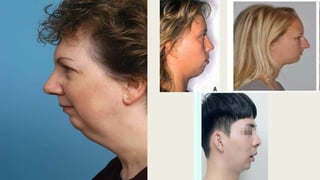
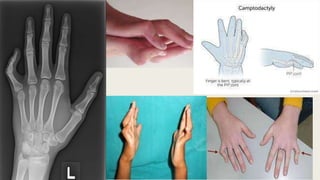
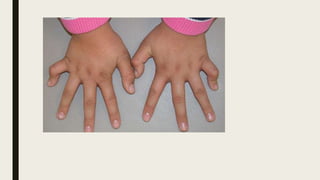
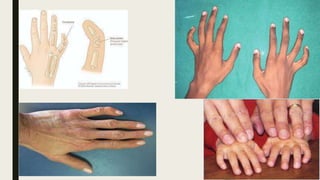
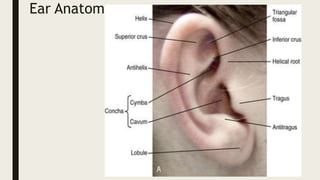
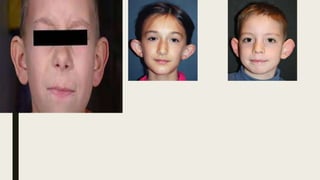
This document provides information on various congenital hand deformities, including camptodactyly, clinodactyly, and prominent ears. Camptodactyly is a flexion deformity of the PIP joint that most commonly affects the little finger. It is classified into three types based on presentation and severity. Clinodactyly is a deviation of the digit in the radial or ulnar direction, often affecting the small finger. Prominent ears are characterized by an abnormally protruding pinna due to underdeveloped antihelix and concha. Management involves splinting or surgery depending on severity and age of presentation.
















![Management
■ Prior to 6 months of age, the ear cartilage is very soft and may be amenable to
moulding and splinting. Bandaging and taping have been used in the past but
now sophisticated splints have been designed to correct problems more
specifically[3].
■ Ear splintage can be a very effective technique for treatment of neonates with
deformational auricular anomalies. After 6 months, surgical correction is the
only option.](https://image.slidesharecdn.com/campto-221008123248-8d4e4454/85/CAMPTODACTYLY-ppt-21-320.jpg)