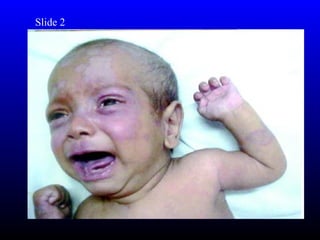
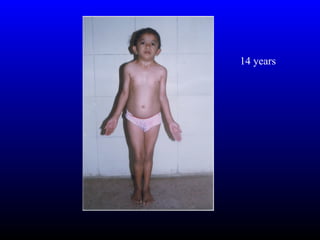
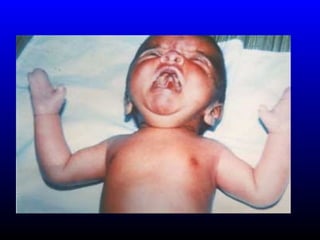
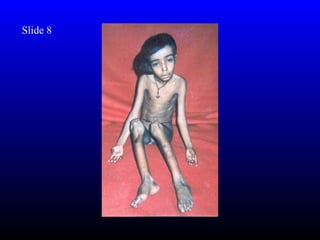
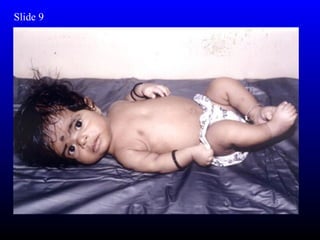
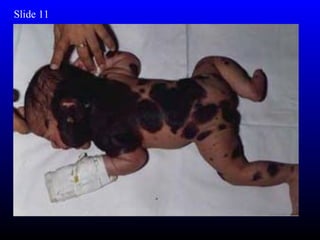
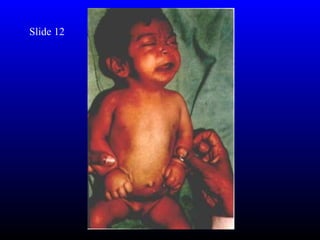
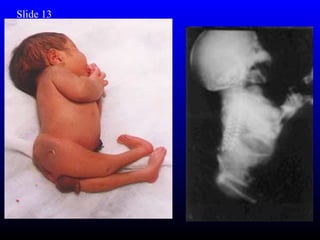
The diagnosis is Turner syndrome. Some key risk factors include:
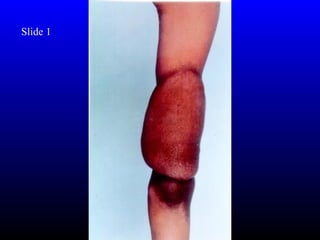
- Cardiac anomalies like bicuspid aortic valve and coarctation of the aorta, present in around 50% and 20% respectively. Early detection and management is important.
- Hypothyroidism, present in 10-30% of patients, requiring lifelong thyroid supplementation.
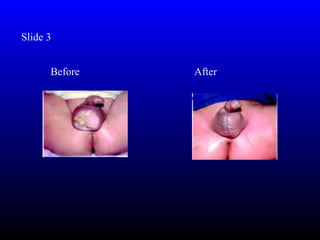
- Infertility due to gonadal dysgenesis. Spontaneous puberty and menses occur in only 20-25% and 2-5% respectively. Assisted reproductive technology may be the only option for biological children.
- Increased risk of diabetes, hypertension and metabolic syndrome later in life requiring lifestyle management and treatment.
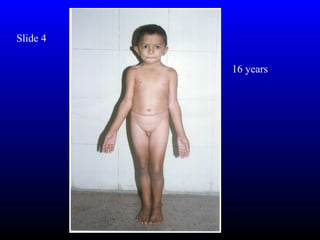
- Reduced linear growth



























































































![[Mary sheridan] from_birth_to_five_years_children(bookos.org)[1]](https://cdn.slidesharecdn.com/ss_thumbnails/marysheridanfrombirthtofiveyearschildrenbookos-200719030205-thumbnail.jpg?width=640&height=640&fit=bounds)



























