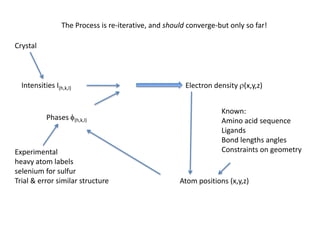
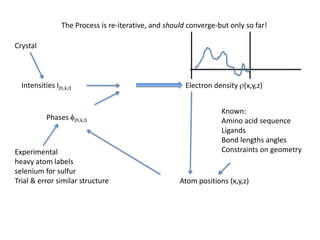
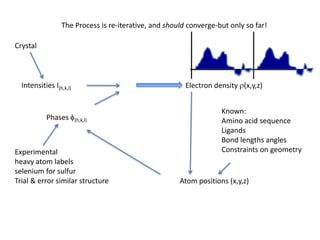
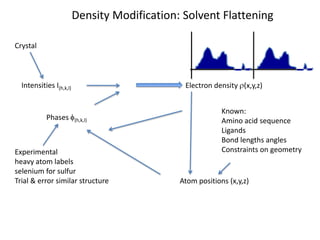
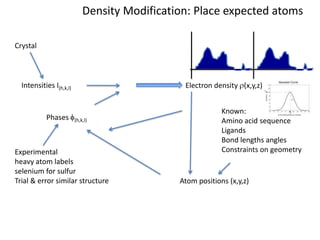
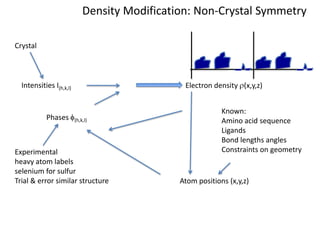
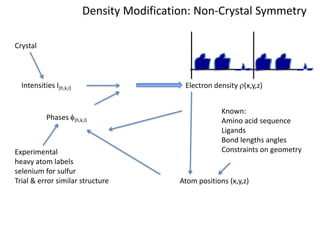
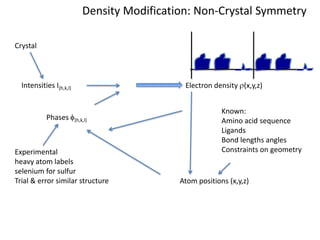
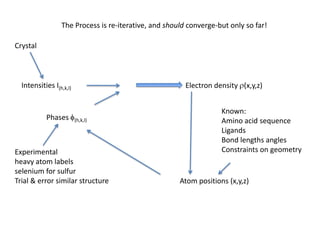
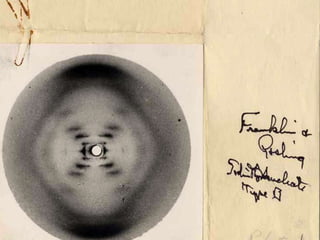
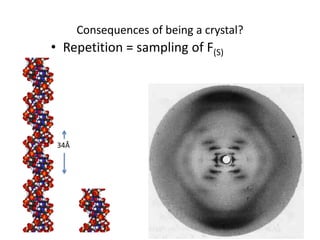
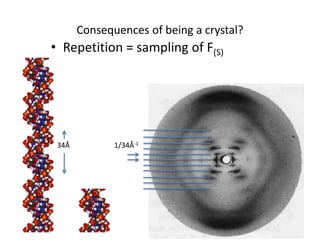
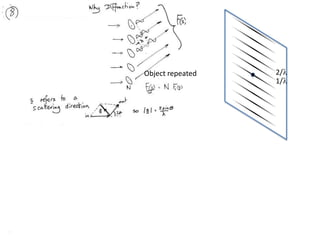
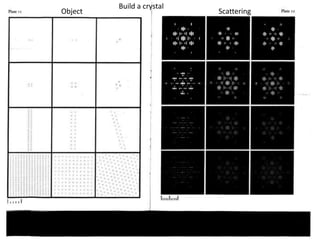
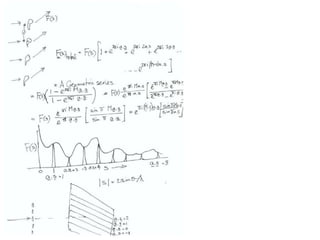
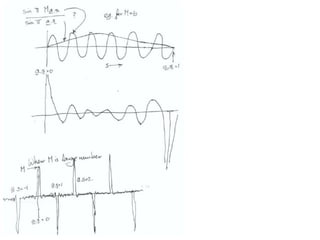
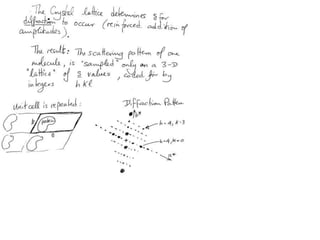
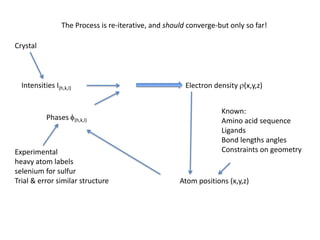
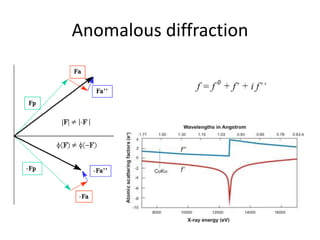
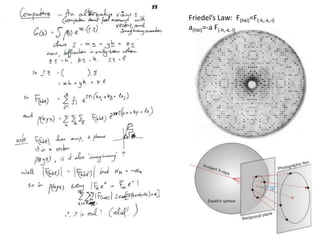
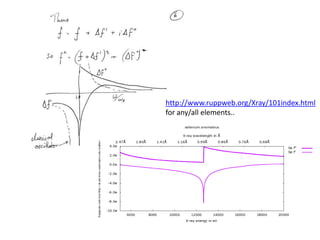
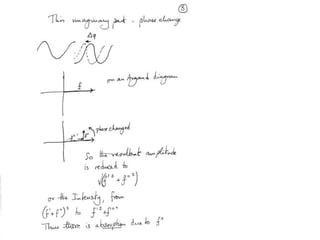
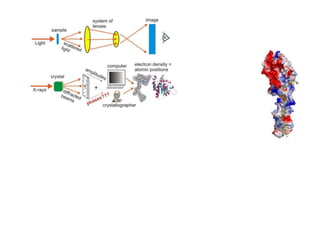
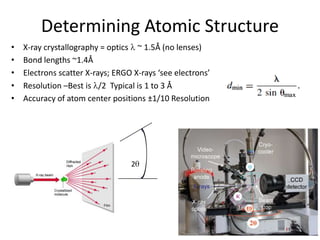
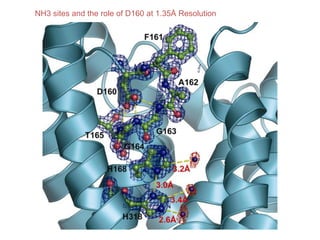
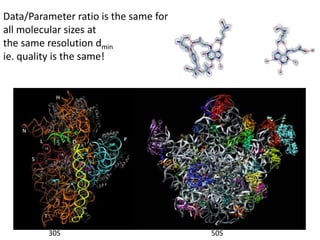
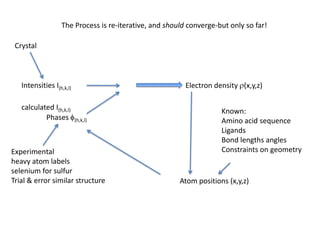
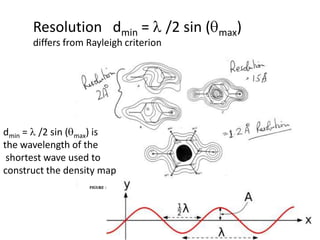
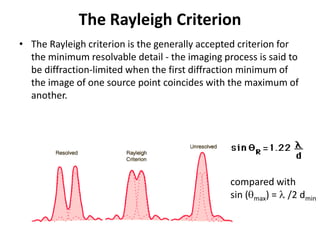
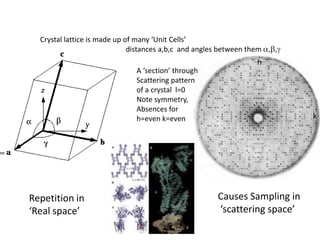
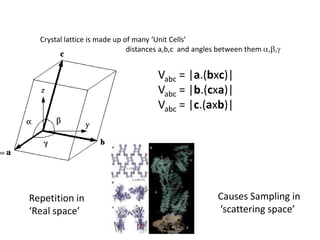
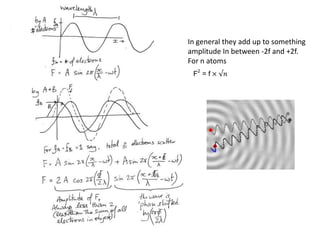
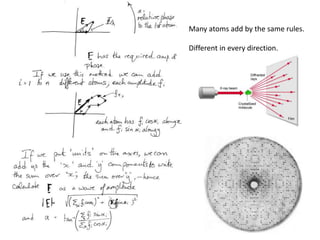
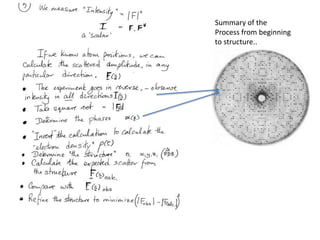
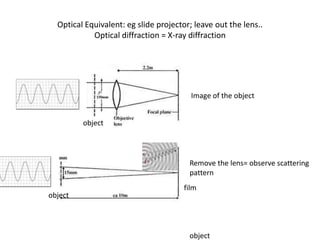
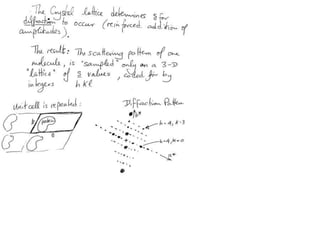
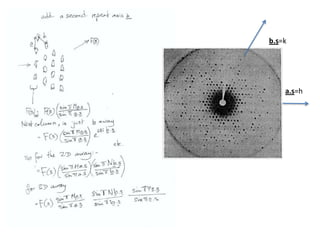
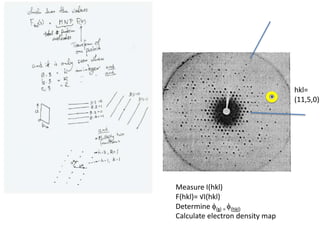
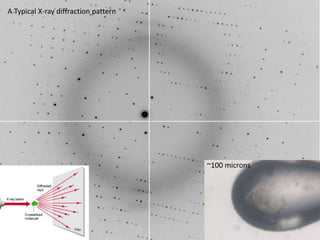
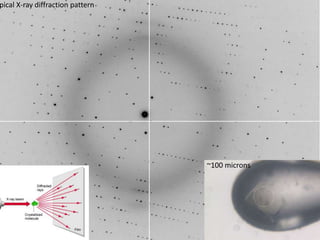
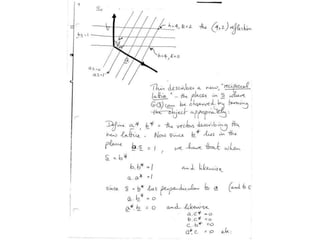
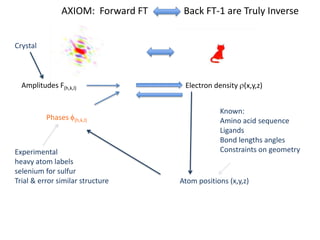
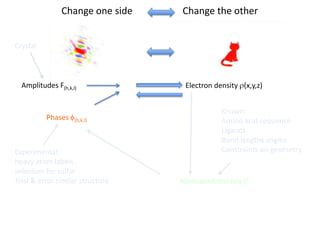
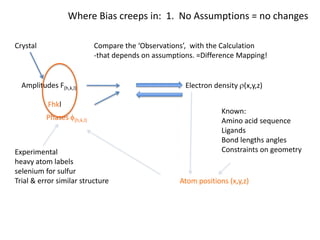
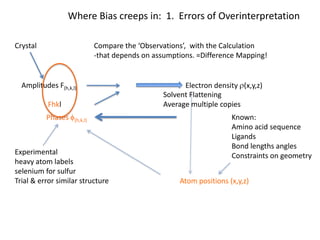
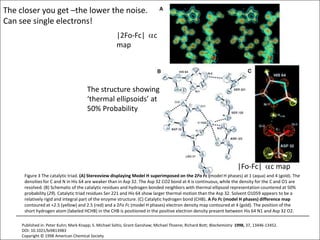
This document provides resources and an overview of topics for a course on crystallography and protein structure determination using X-ray crystallography. The course will involve crystallizing a protein, collecting data at the Advanced Light Source, and determining the protein's atomic structure. Key topics covered include X-ray scattering, the phase problem, structure refinement, and sources of errors. Online resources and contacts are provided for computing, tutorials, and beamline information.









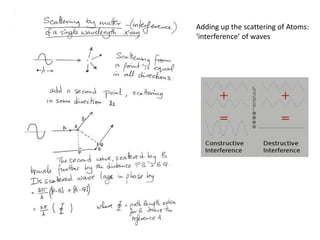
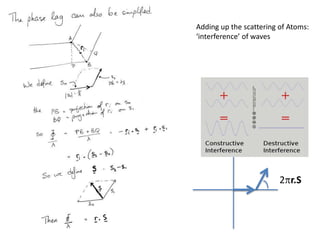
![ScatteringAdding up the scattering of Atoms:‘interference’ of wavesWaves add out of phaseby 2p[extra path/l]](https://image.slidesharecdn.com/bp219-2011-4-13-110419122135-phpapp01/85/Bp219-2011-22-320.jpg)



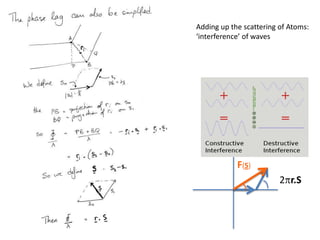

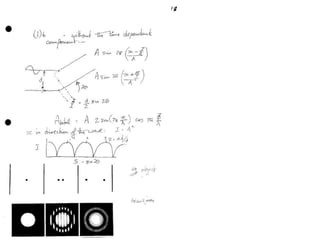









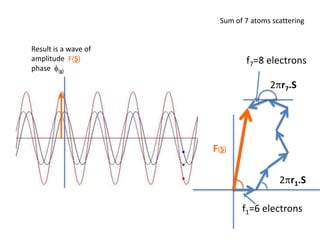
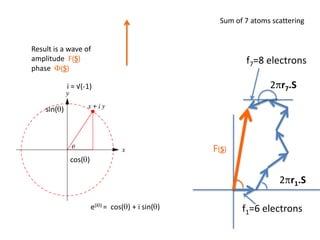
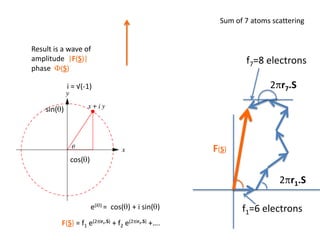
![Argand Diagram.. F(S) = |F(s)| eiqIntensity = |F(s)|2How to represent I(s)?I(s) = |F(s)|2 =F(S) .F*(S) proof?Where F*(S) is defined to be the ‘complex conjugate’ of F(S) = |F(s)| e-iqso |F(s)|2 =|F(s)|[cos(q) + isin(q)].|F(s)|[cos(q) - isin(q)]=|F(s)|2 [cos2 (q) + sin2 (q)]=|F(s)|2 R.T.P.F(S)2pr.Sq](https://image.slidesharecdn.com/bp219-2011-4-13-110419122135-phpapp01/85/Bp219-2011-46-320.jpg)



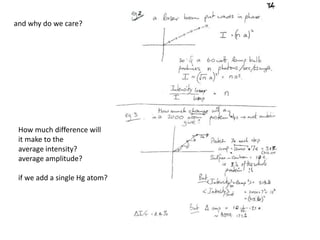


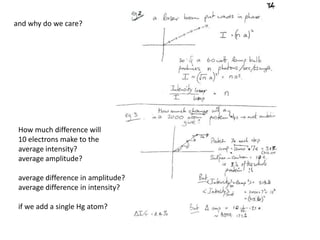
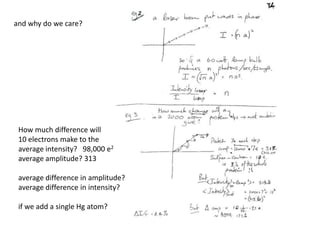
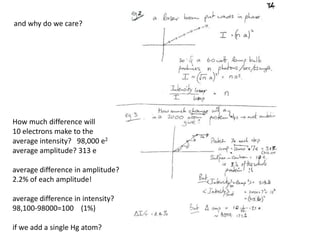







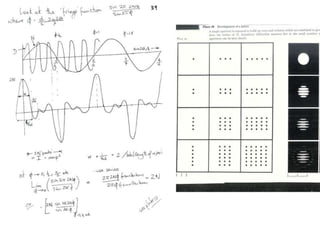

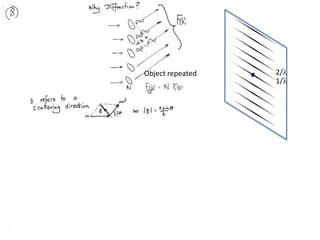

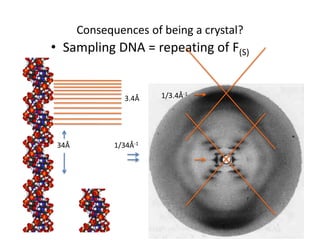

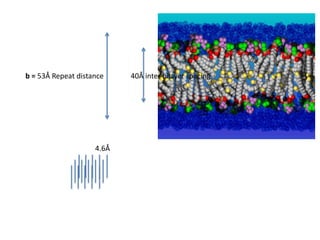
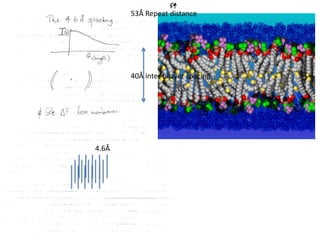
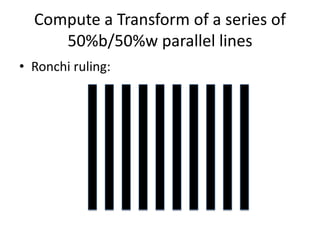
![Transform of two hoizontal lines defined y= ± y1F(s) = Int [x=0-infexp{2pi(xa+y1b).s} + exp{2pi(xa-y1b).s}dVr ]=2 cos (2py1b).s * Int [x=0-infexp{2pi(xa).s dVr]for a.s=0 the int[x=0-infexp{2pi(xa).s dVr] = total e content of the linefor a.s≠0 int[x=0-infexp{2pi(xa).s dVr] = 0hence r(r)isa line at a.s= 0 parallel to b, with F(s) = 2 cos (2py1b).s -along a vertical line perpendicular to the horizontal lines.This structure repeats along the b direction thus peaks occur at b.s= k (where k is integer only)Transform of a bilayer..b=53Å](https://image.slidesharecdn.com/bp219-2011-4-13-110419122135-phpapp01/85/Bp219-2011-73-320.jpg)




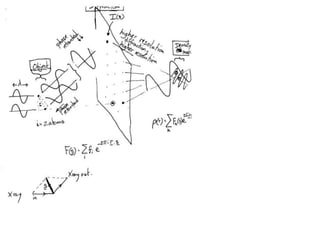


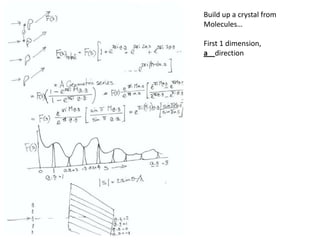
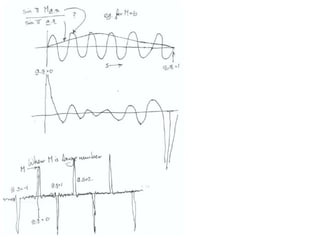


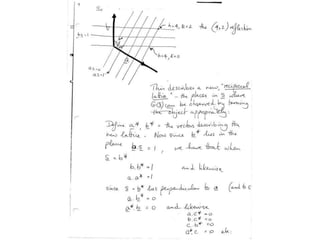

















![If a map is produced with some f(hkl)The probability of it being correct is P(hkl)P(hkl) (f(hkl))Maximum value of P(hkl) (f(hkl)) gives the ‘Most probable’ mapMap with the least mean square error, is when noise is minimum, Int find f(best) such that Q= Intf [|F| P(hkl) (f(hkl))exp (if(hkl)) - Fbestf(best))]2df is minimum.is minimum when dQ/dFbest= 0so Fbestf(best) = f |F| P(hkl) (f(hkl))exp (if(hkl))dfFbestf(best) = m|F| center of ‘mass’ of the Probability distribution m = IntP(hkl) (f(hkl))cos(f - f(best))consider errors from one reflection, and its complex conjugate<(Dr)2> = 2/V2 Then F= IntFcos(f - f(best))/ FNoise = 1/V Int F sin (f - f(best))/F = F(1-m2)mF where](https://image.slidesharecdn.com/bp219-2011-4-13-110419122135-phpapp01/85/Bp219-2011-120-320.jpg)

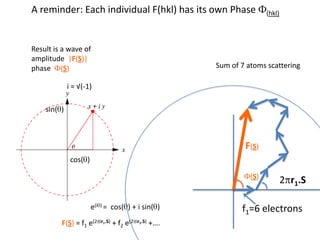
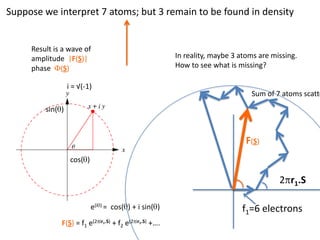
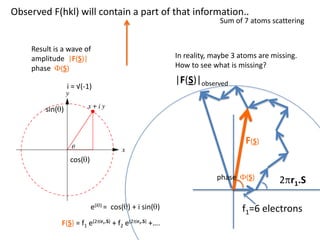
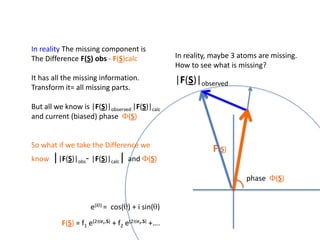
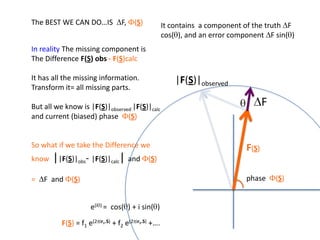
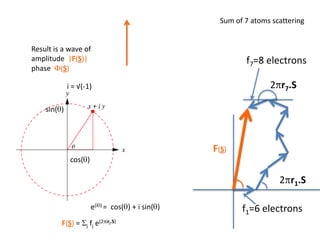
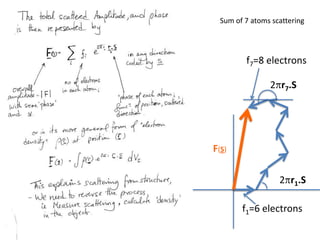
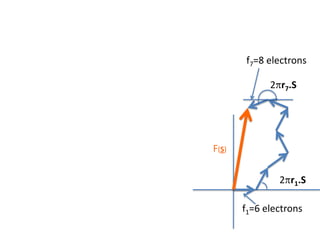
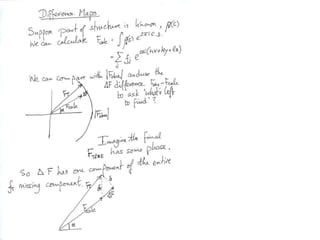
![What contribution to the ‘missing truth’?It contains a component of the truth DF cos(q)and an error component DF sin(q)So what if we transform the Difference weknowDF =||F(S)|obs- |F(S)|calc| and F(S)On average DF = ftruecos(q)so On average towards the truth isDF cos(q) = ftruecos2(q)What is the average value of cos2(q)?= ½ ( cos 2q +1)cos(q)|F(S)|observed2pr7.SDFqftrueF(S)p/2+ 1dq/ 1/2p/22pr1.Sp/2= ½ 0[ -f sin2q + q]/= ½ fie transform DF, F(S) = ½ true density for 3 missing atomsphase F(S)p/2e(iq) = cos(q) + isin(q)F(S) = f1 e(2pir1.S) + f2 e(2pir2.S) +….](https://image.slidesharecdn.com/bp219-2011-4-13-110419122135-phpapp01/85/Bp219-2011-132-320.jpg)

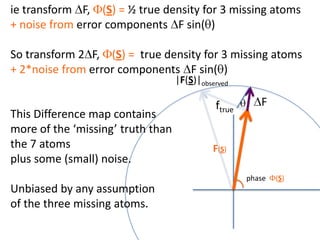
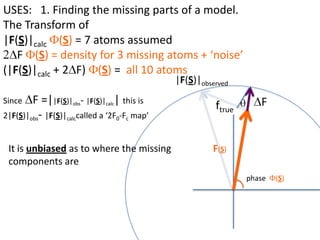
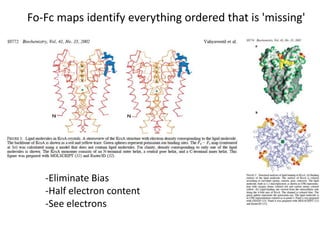
![USES: 2. Add a substrate, Grow a new crystalMeasure New |F(S)|obs+substrateCompare with the apo-protein.TransformDF =||F(S)|obs+substrate- |F(S)|obs|F(S)or[2|F(S)|obs+substrate- |F(S)|obs ] F(S)= a ‘2F0-Fo map’cos(q)|F(S)|observed2pr7.SDFqftrueF(S)It is unbiased as to where the missingsubstrate is.2pr1.Sphase F(S)e(iq) = cos(q) + isin(q)F(S) = f1 e(2pir1.S) + f2 e(2pir2.S) +….](https://image.slidesharecdn.com/bp219-2011-4-13-110419122135-phpapp01/85/Bp219-2011-136-320.jpg)

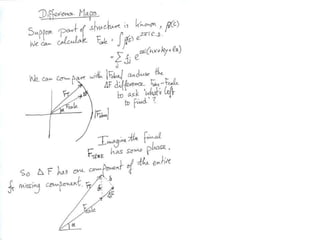





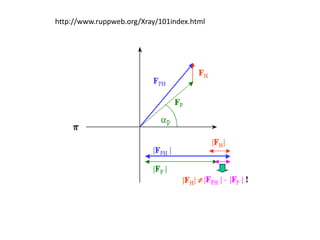
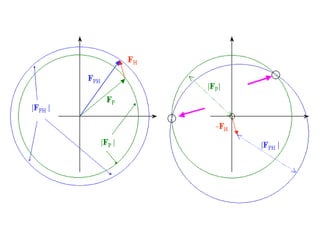
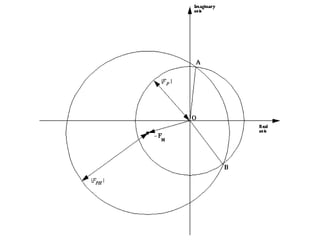

























![Transform of I(hkl) = Transform of F2(hkl)…What happens if I transform what I do know and can measure, namely I(S) = |F(S)|observedieP(r) = S|F(S)|2e(-2pir.S)= S [F(S).F*(S)]e(-2pir.S) ?](https://image.slidesharecdn.com/bp219-2011-4-13-110419122135-phpapp01/85/Bp219-2011-179-320.jpg)