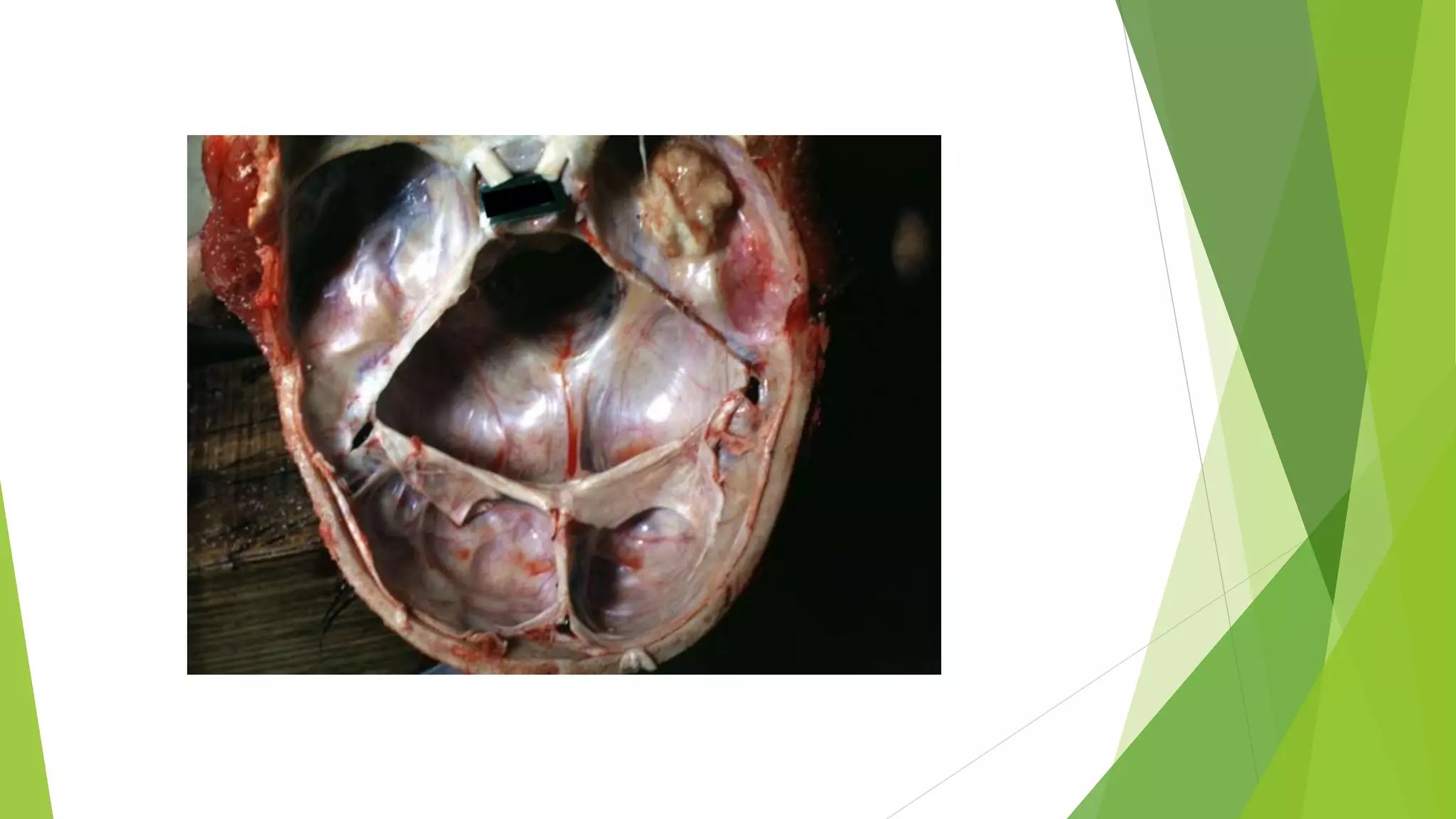
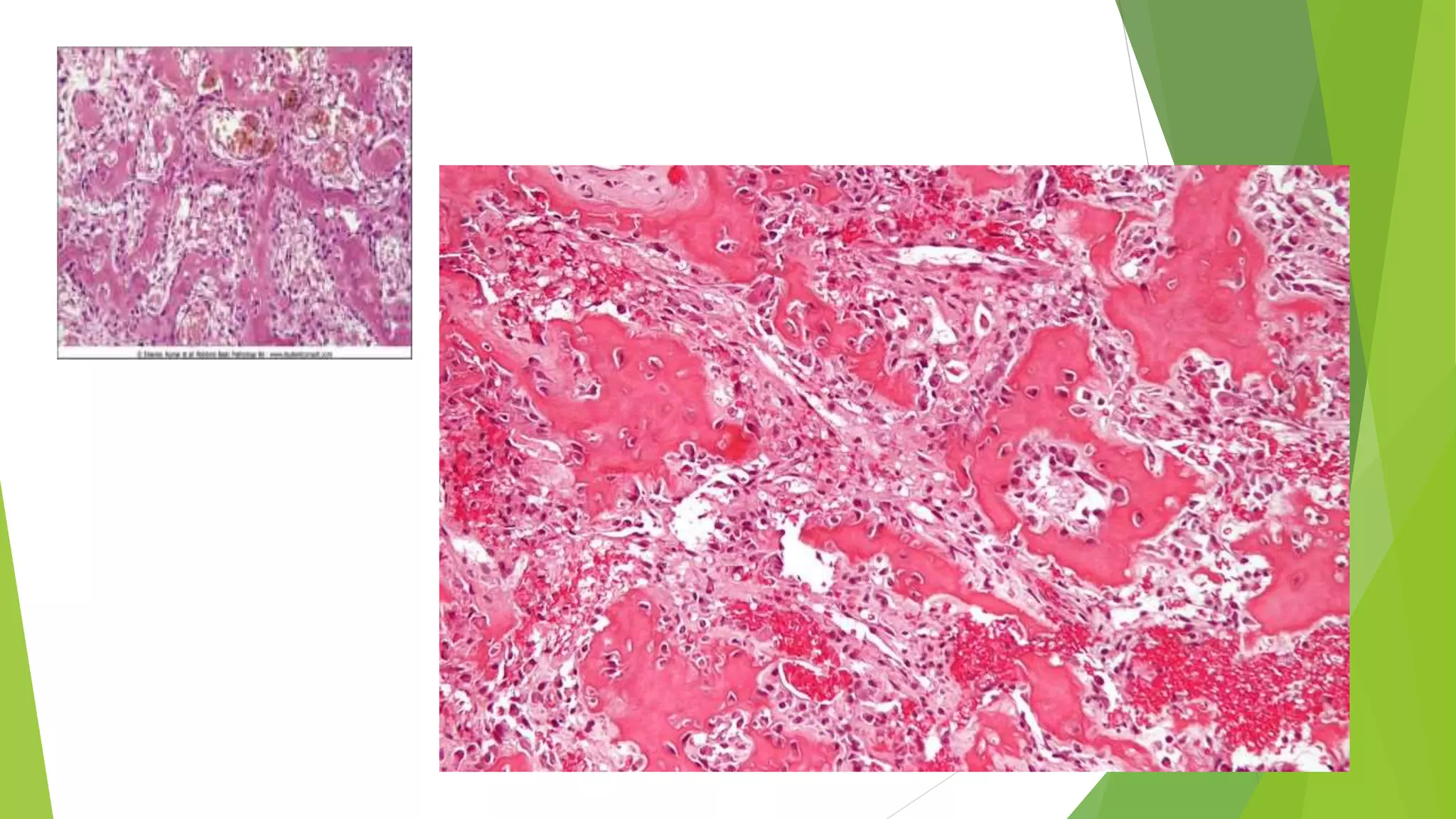
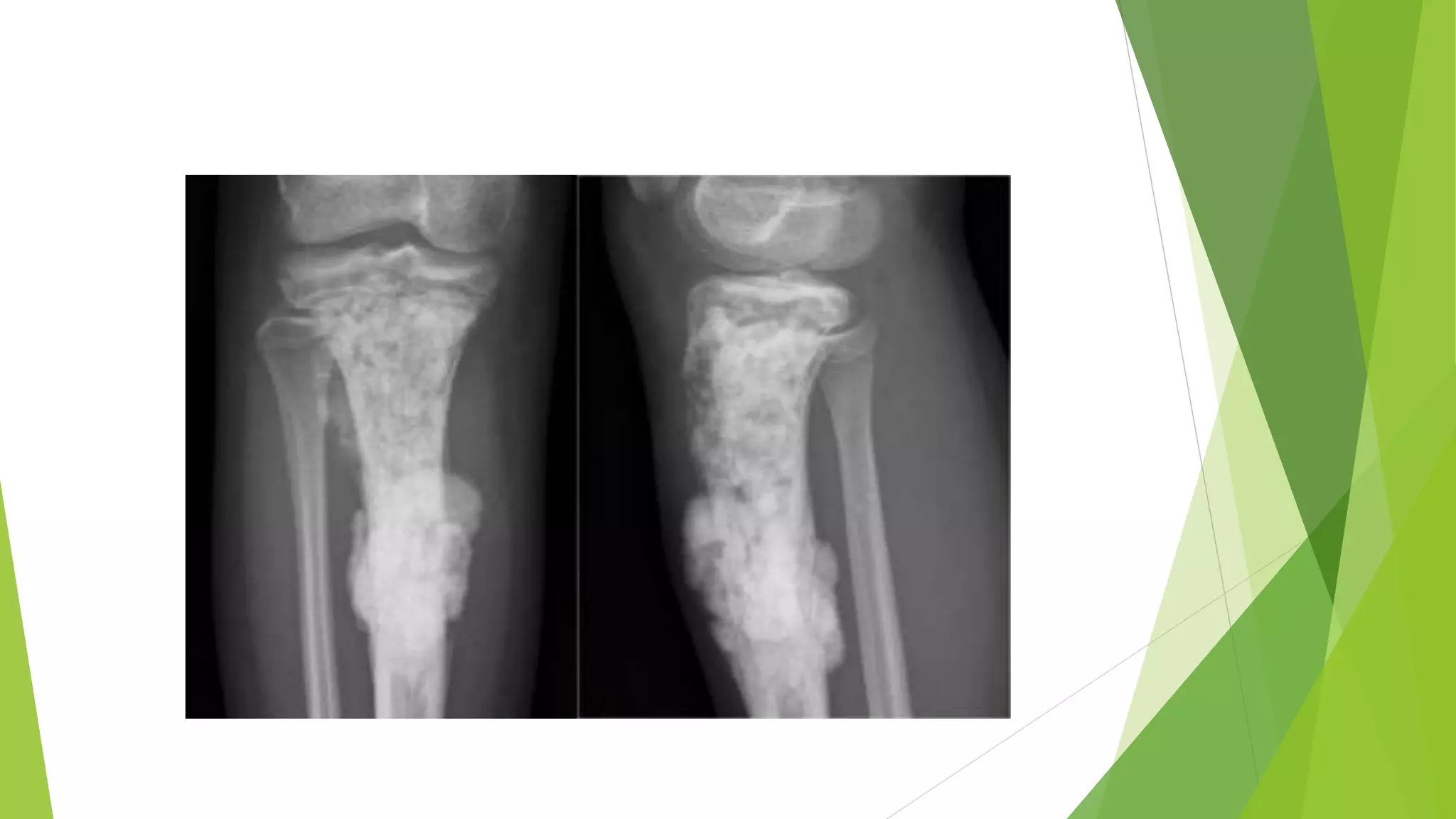
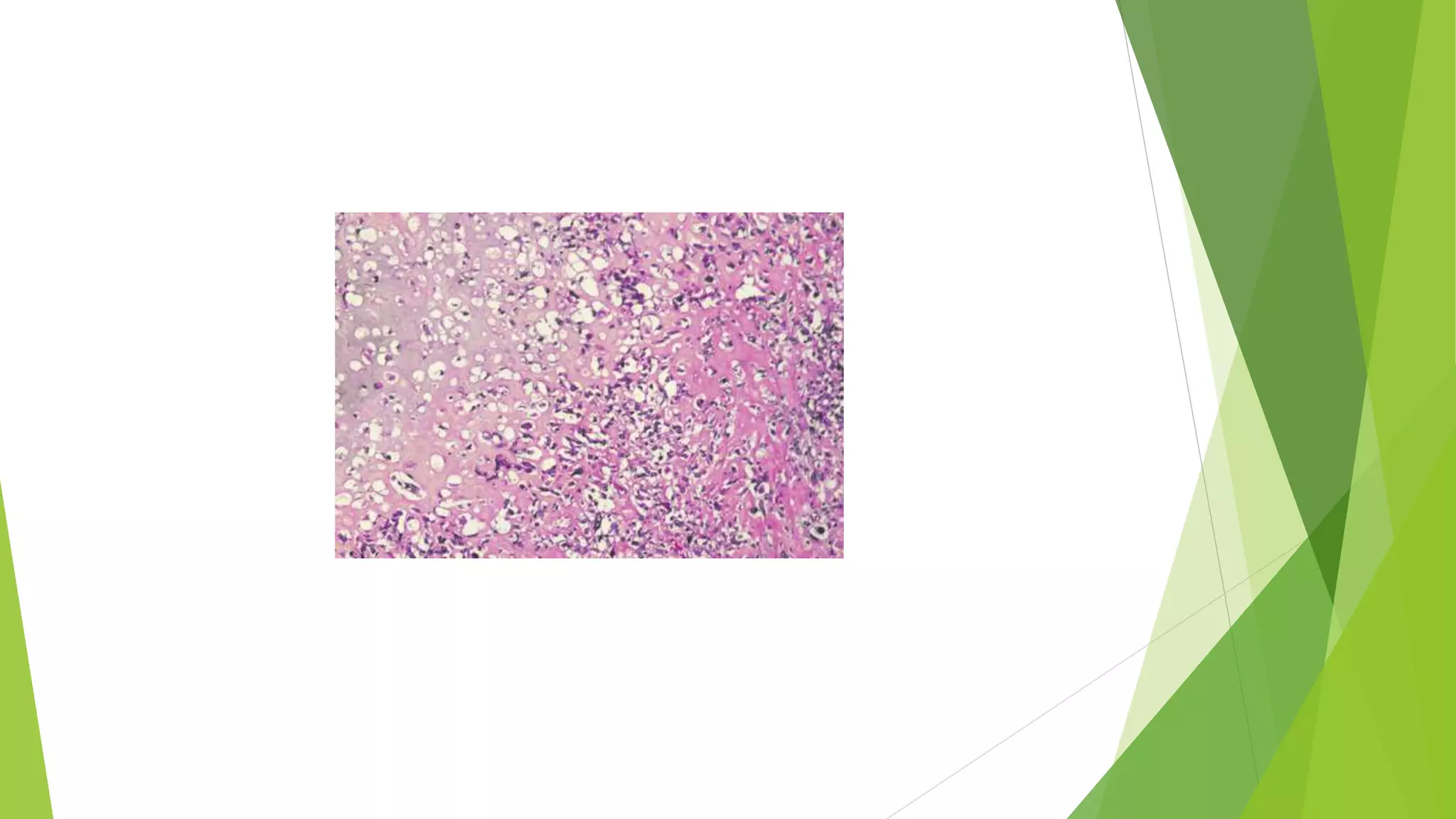
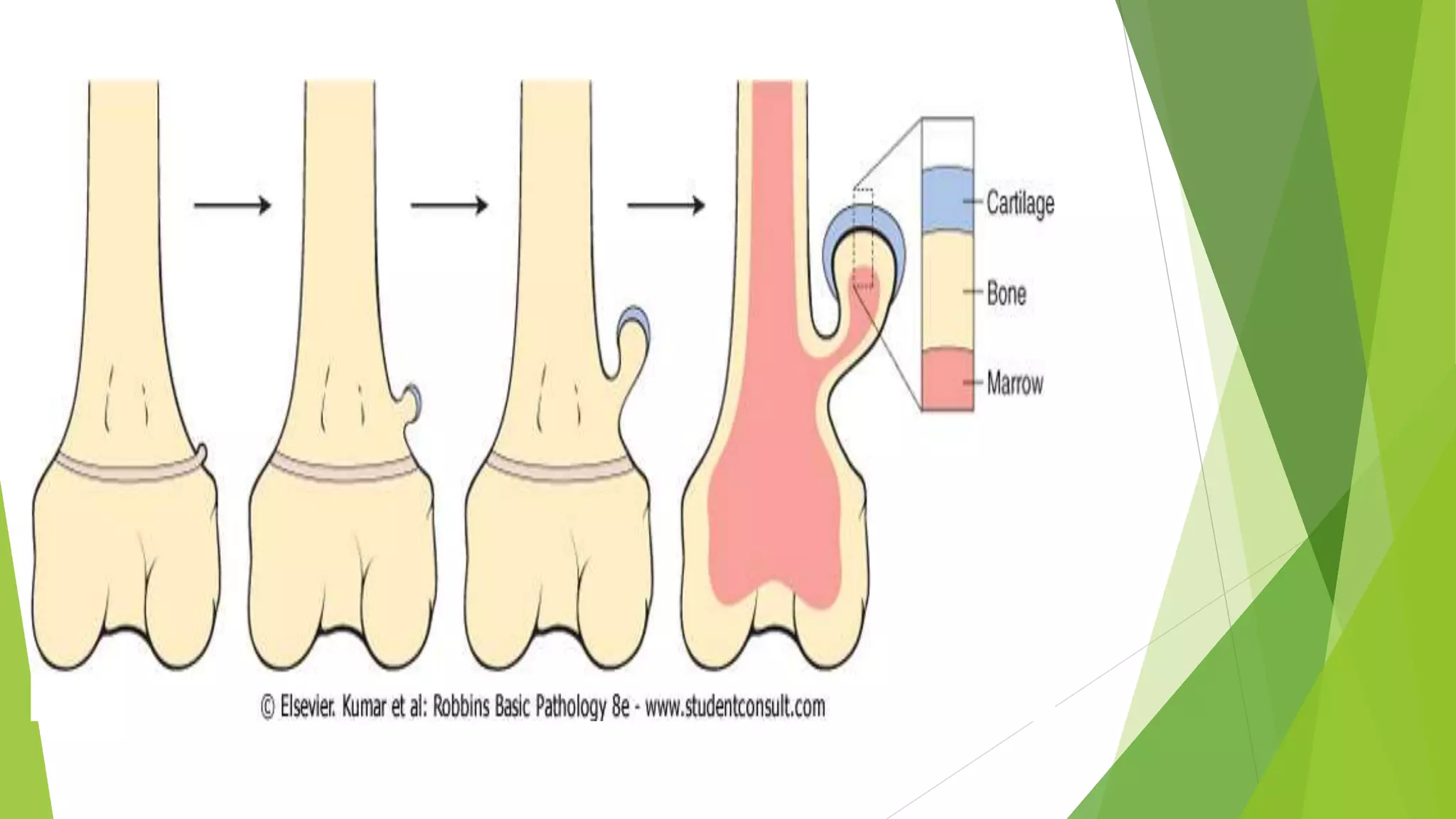
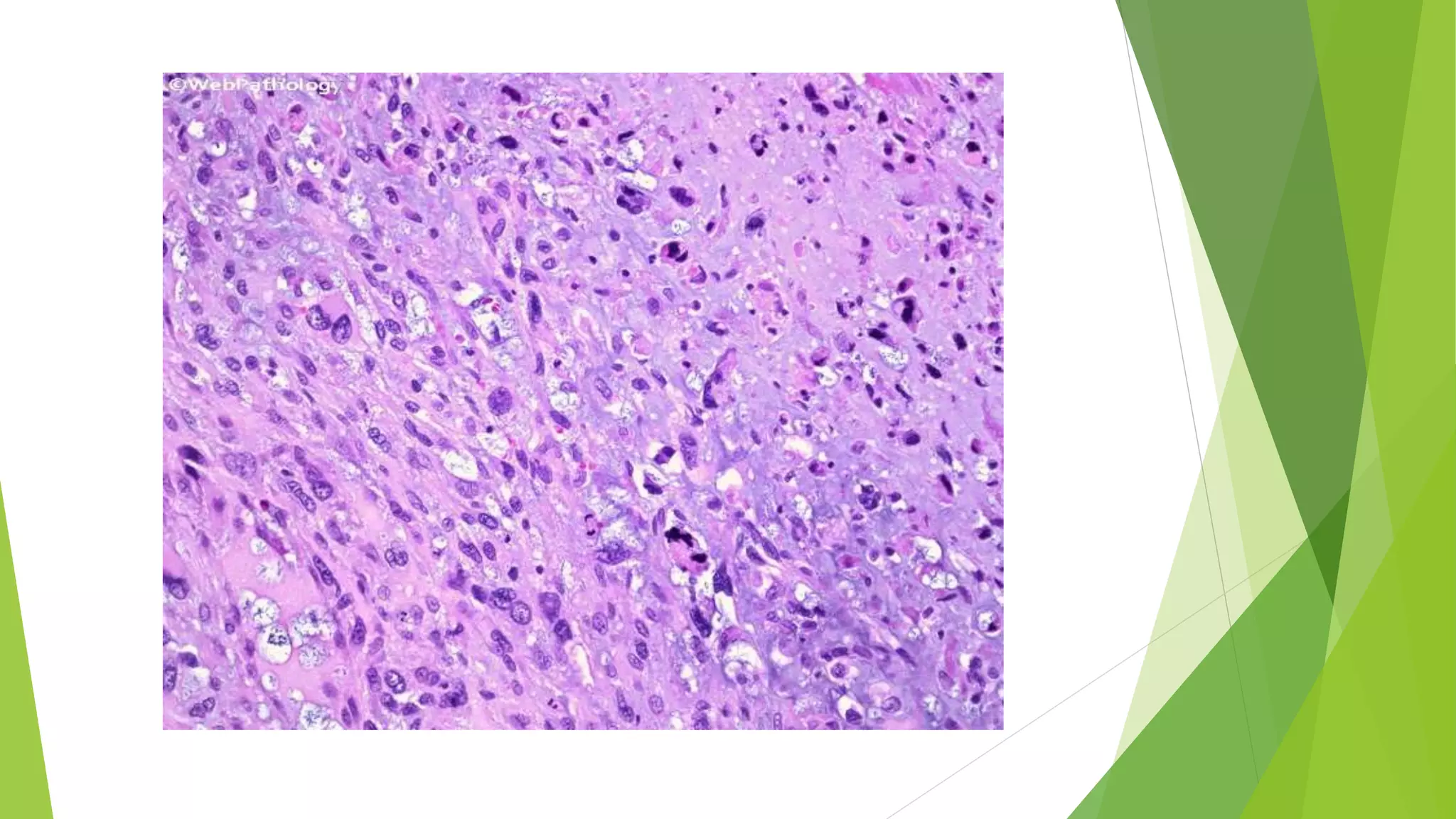
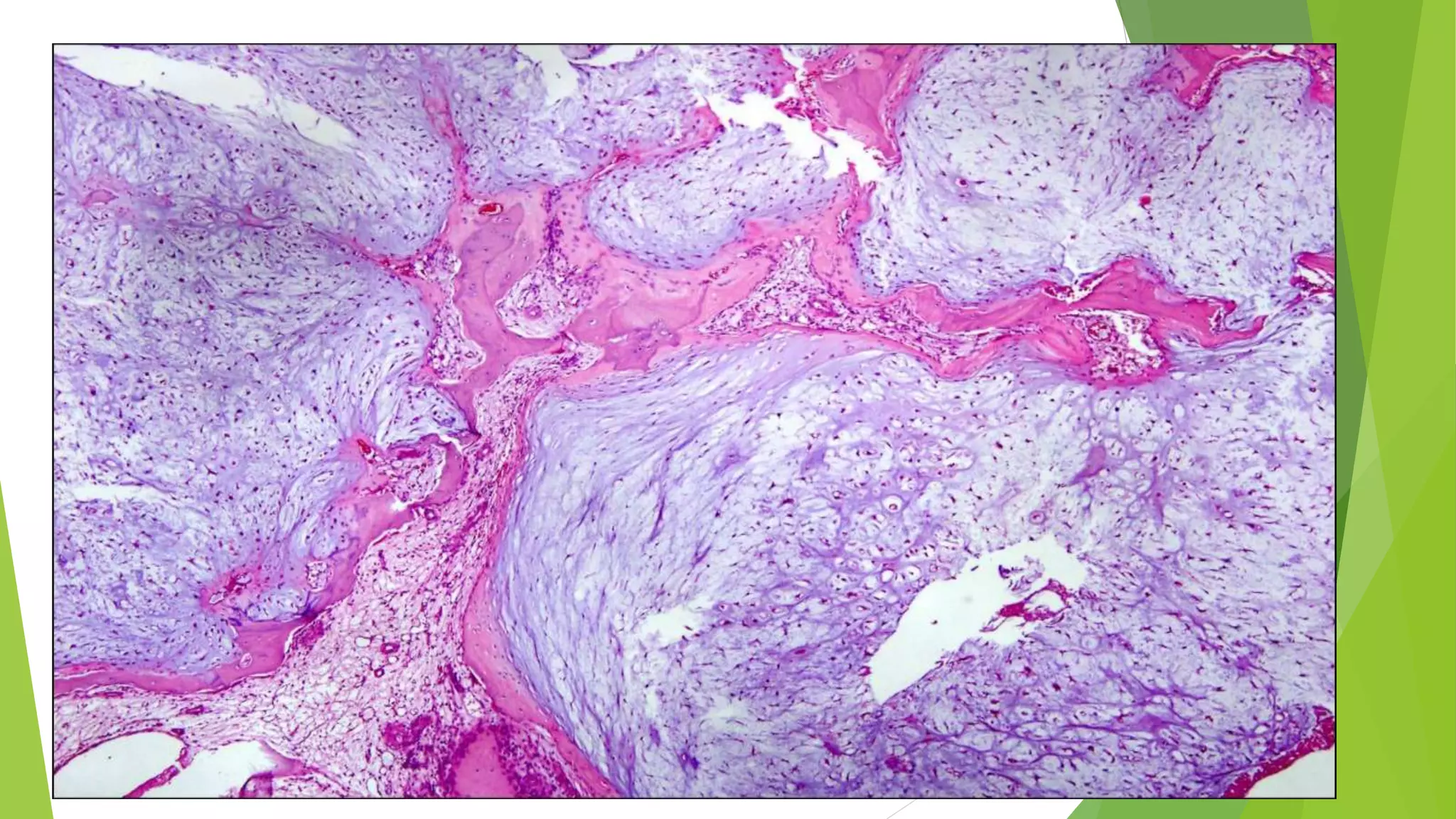
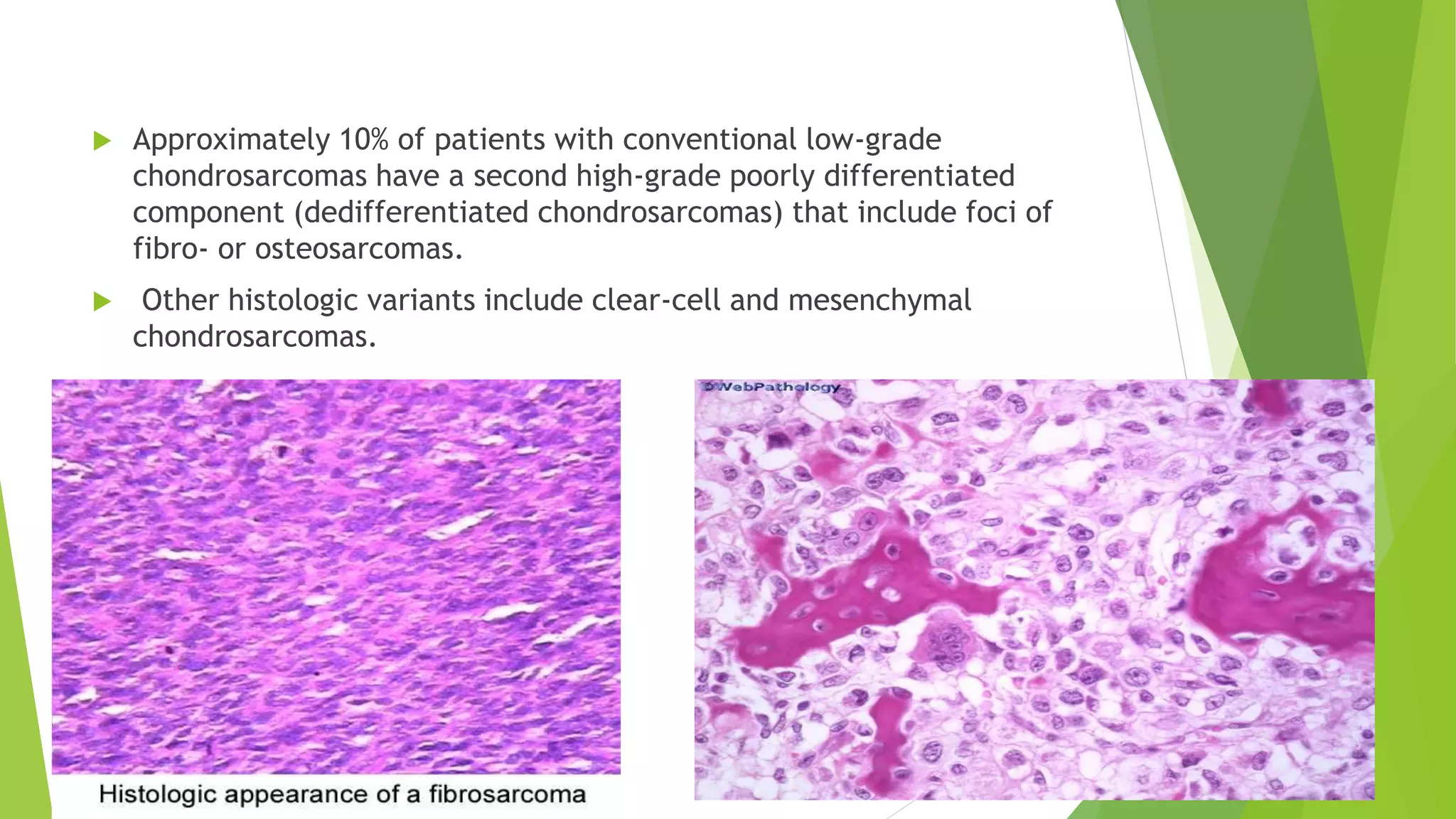
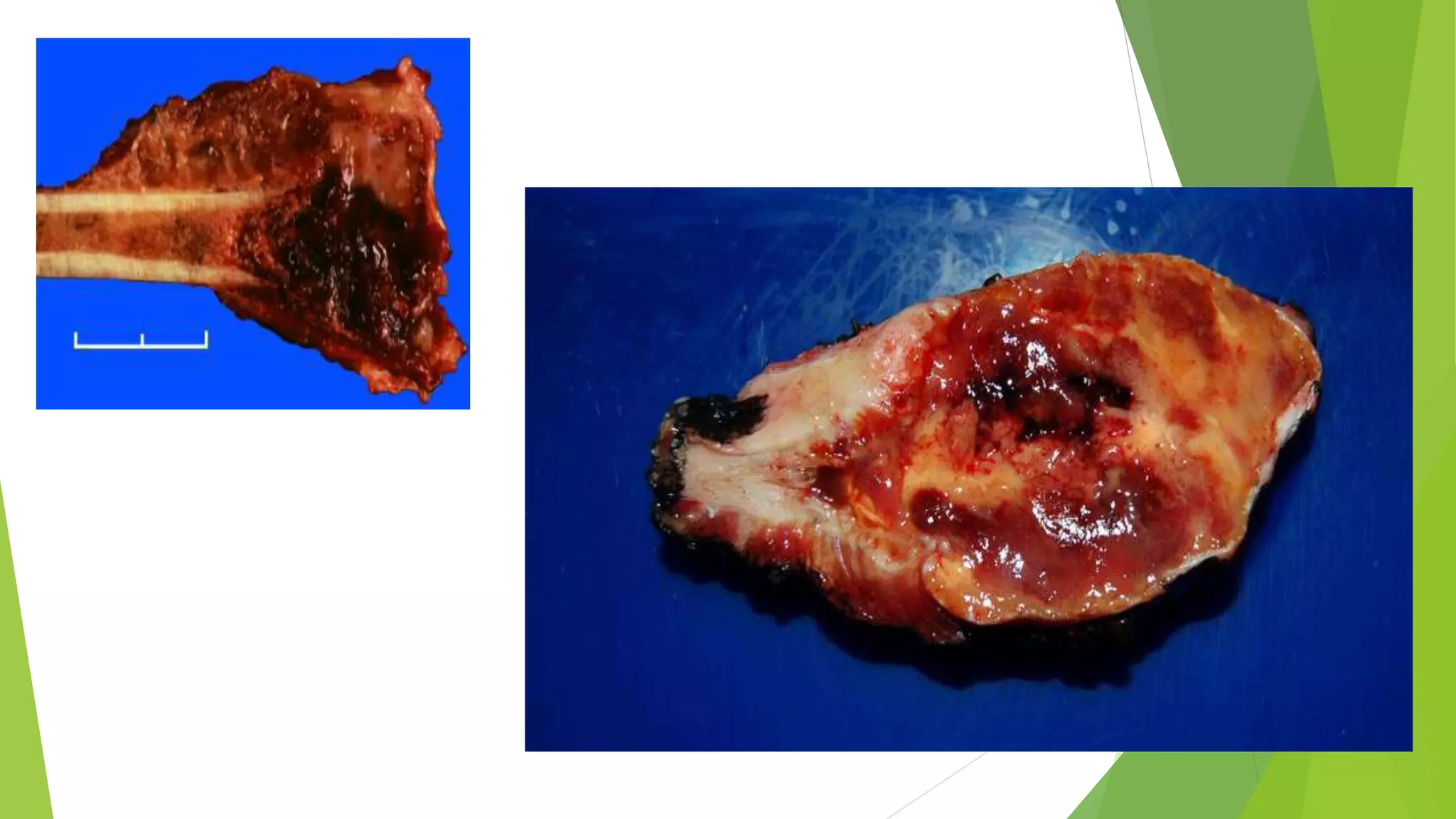
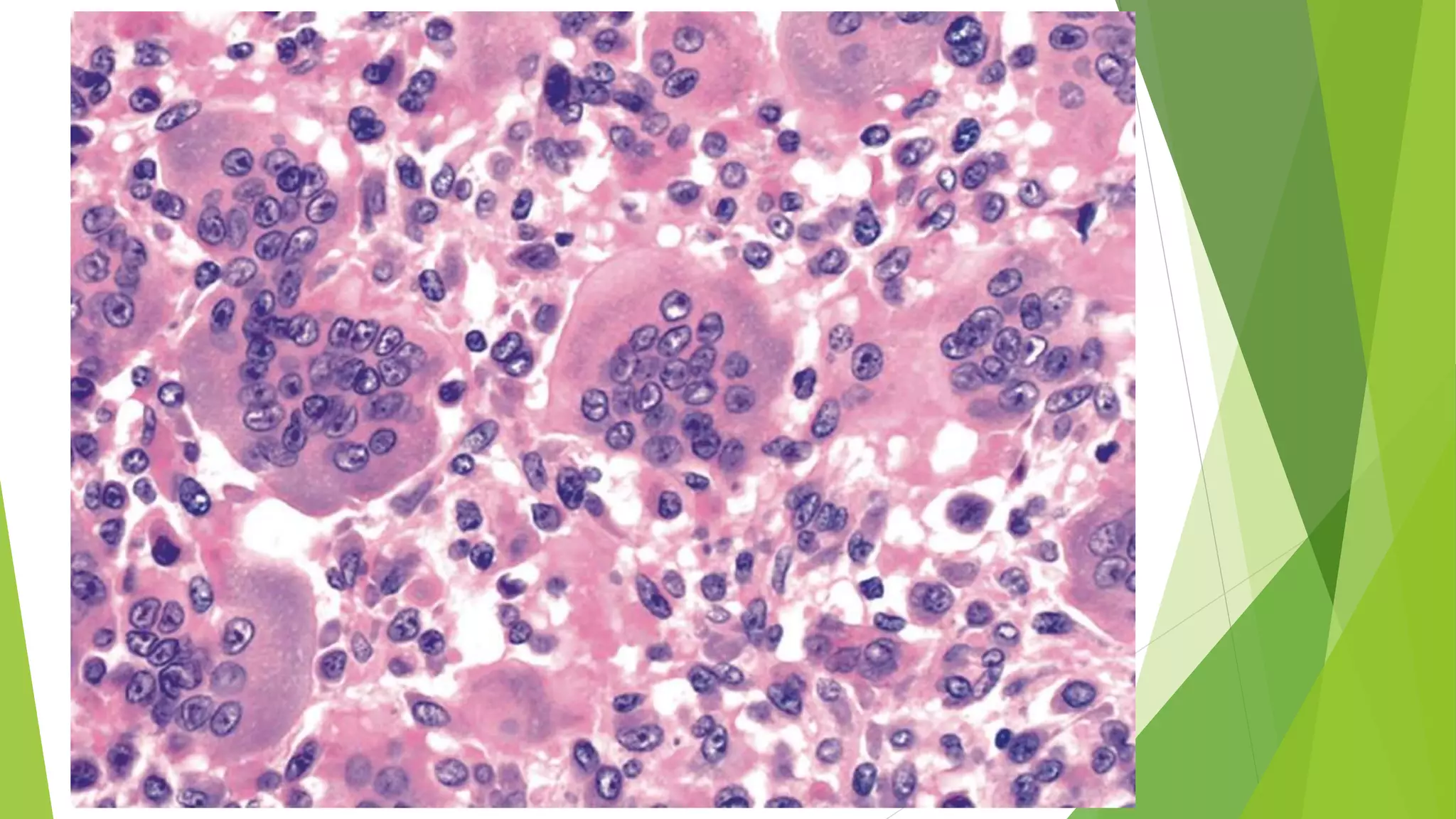
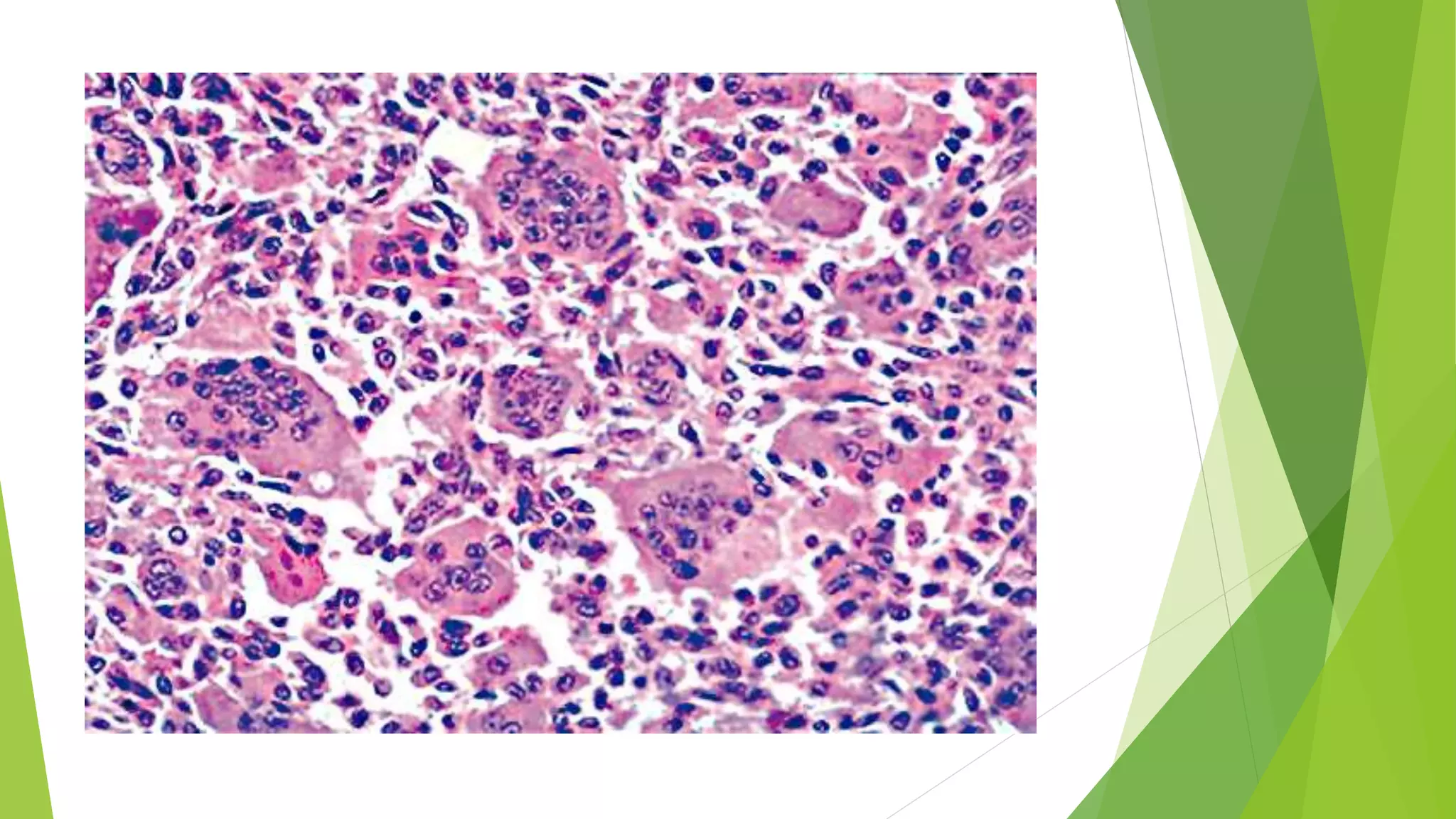
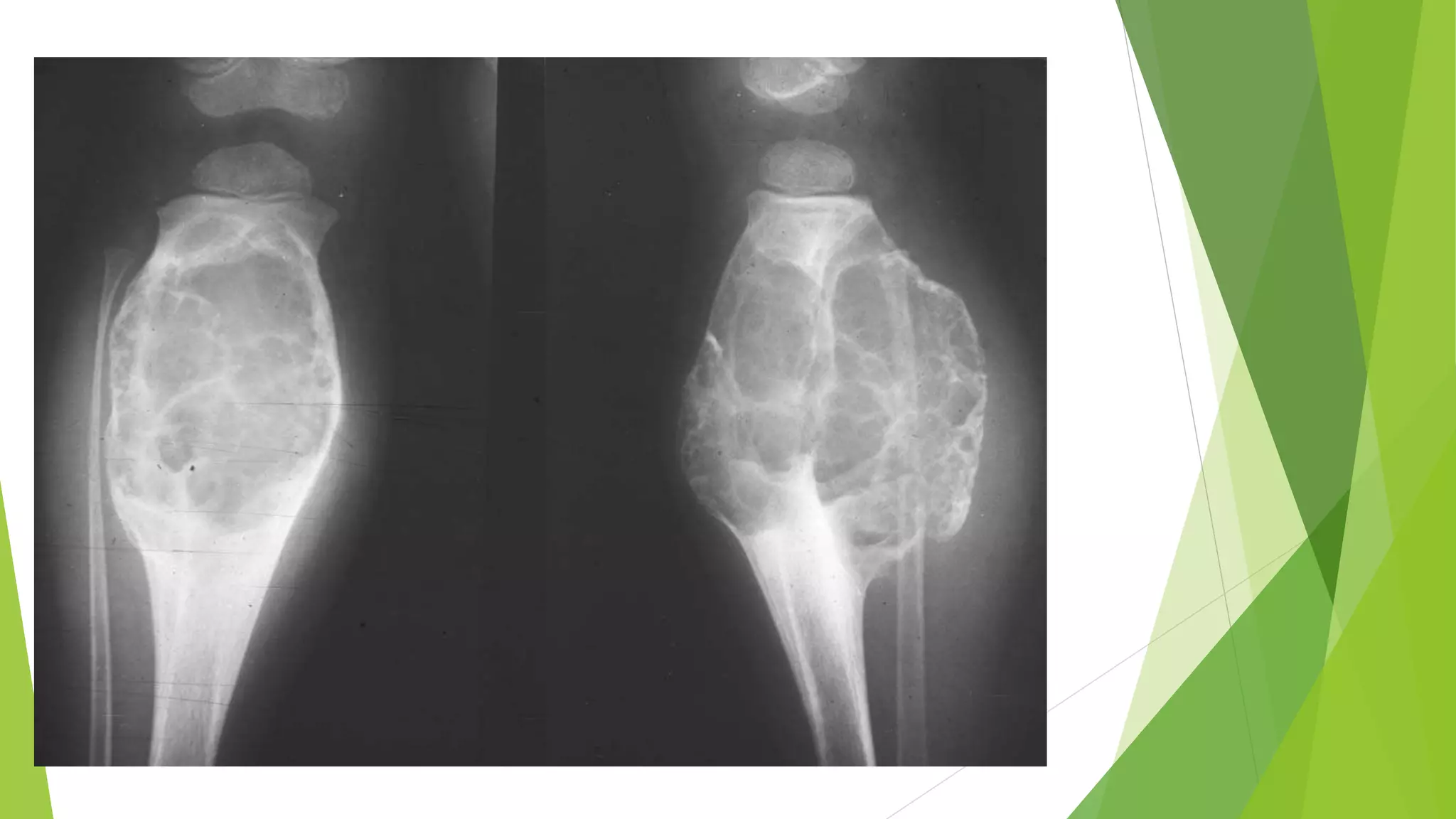
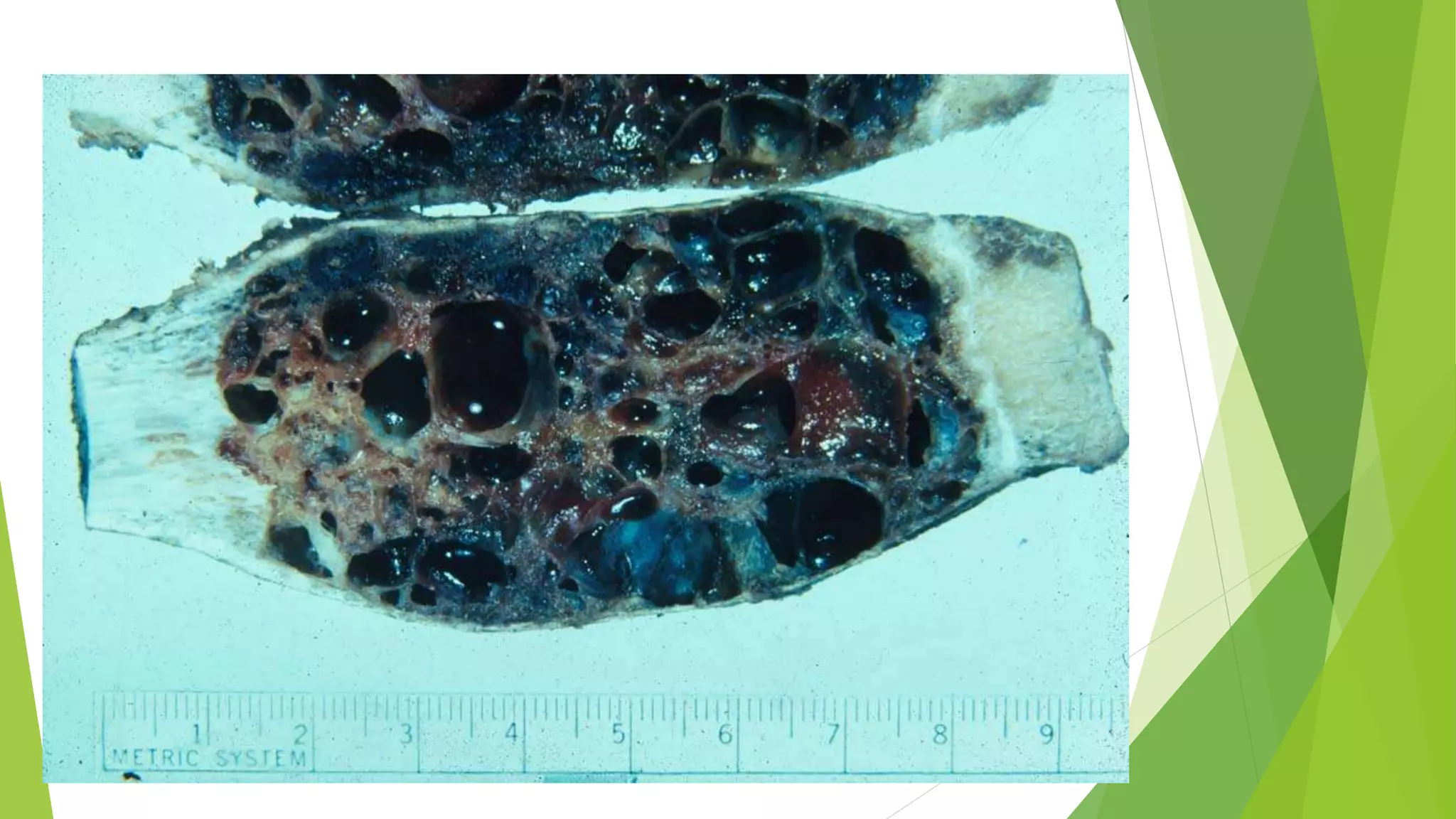
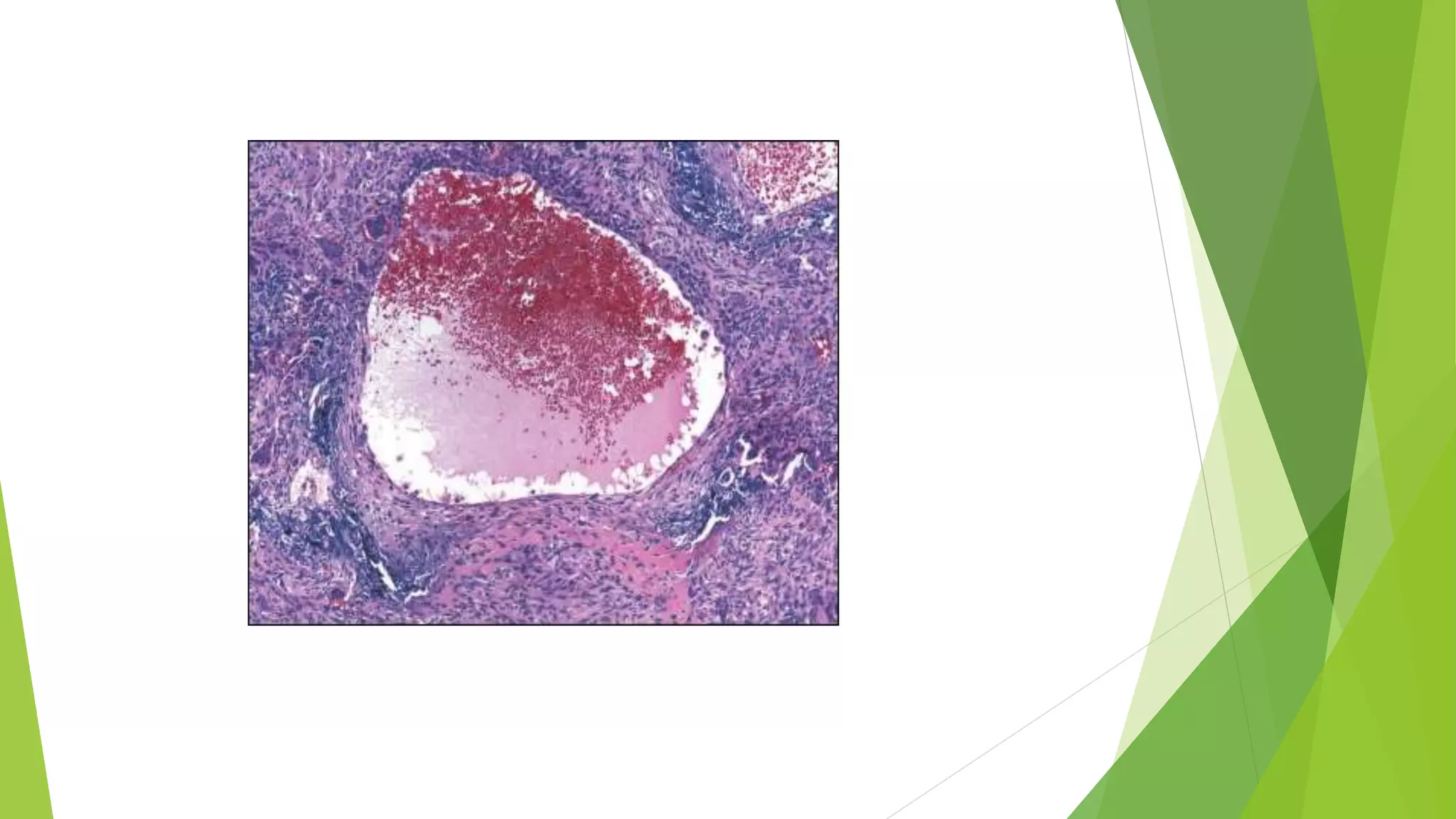
Bone tumors can develop at any age and in various locations. Most osteosarcomas occur in adolescents around the knee, while chondrosarcomas tend to develop in mid-to-late adulthood in the trunk and proximal long bones. Bone tumors may be benign like osteomas, osteoid osteomas, or osteoblastomas, or malignant like osteosarcomas. Osteosarcomas are the most common primary bone cancer and often present as painful masses, usually metastasizing to the lungs. Ewing sarcoma and primitive neuroectodermal tumors are small round cell tumors that predominantly affect children and young adults.