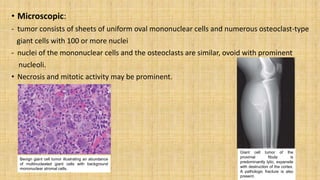
This document provides information on various bone tumors. It begins by defining tumors and distinguishing between benign and malignant types. It then describes the four main cell types that make up bone tissue and their functions. Various bone tumors are classified based on the sites they occur and whether they form bone or cartilage. Specific bone forming tumors discussed include osteoid osteoma, osteoblastoma, and osteosarcoma. Cartilage forming tumors described are osteochondroma, chondromas, and chondrosarcoma. Other tumors discussed are Ewing sarcoma and giant cell tumor. For each tumor, the document outlines characteristics such as affected age groups, common sites, signs/symptoms, pathogenesis, morphology, microscopy images, and treatment approaches.